
{ DOWNLOAD AS PDF }
ABOUT AUTHORS
Suleman S. khoja * 1, Dr. L J. Patel 2, Sohil S. Khoja 3, Karim R. Panjwani 3, Jagdish Ray3
1 PhD. Research Scholar, Ganpat University, Mehesana, Gujarat, India
2 Faculty of Pharmacy, Ganpat University, Mehesana, Gujarat, India
3 Resource person, Pharmaceutical Quality Assurance, Audit and Compliance, Vapi, Gujarat, India
ABSTRACT
Good documentation constitutes an essential part of the quality assurance system and is key to operating in compliance with GMP requirements. Documentation requirements of maintaining complete, accurate, truthful and verifiable data in all cGXP documents that are needed to be maintained as per regulatory requirements and various Governmental regulations, laws, rules and statutes/acts. Documentation may exist in a variety of forms, including paper-based, electronic or photographic media. Ensure that the Document should be free from error and during any point if error identify then rectify with proper reason for correcting including sign and date. A system should be in place to indicate special observations and any changes to critical data. GMP Document must have predefined retention period and document must be stored in secure and easy to retrieve or easily available as and when required. Batch Processing and Packaging Instruction must be in place and Contemporaneous entry provision must be available. Instruction and procedure for using equipment; instrument must be clear and specific. Specification with authorization should be available for analysis of Raw Material and Packing Material, Intermediate and Bulk Product, Finished Product.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-2634
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 7, Issue 01 Received On: 11/11/2018; Accepted On: 28/11/2018; Published On: 01/01/2019 How to cite this article: khoja, S., Patel, L., khoja, S., Panjwani, K. and Ray, J. 2019. Review on Good Documentation practice in pharmaceutical manufacturing unit as per European Union GMP Chapter-4 on Documentation. PharmaTutor. 7, 1 (Jan. 2019), 1-4. DOI:https://doi.org/10.29161/PT.v7.i1.2019.1 |
INTRODUCTION:
• Good documentation constitutes an essential part of the quality assurance system and is key to operating in compliance with GMP requirements.
• Documentation may exist in a variety of forms, including paper-based, electronic or photographic media
• Documentation requirements of maintaining complete, accurate, truthful and verifiable data in all cGXP documents that are needed to be maintained as per regulatory requirements and various Governmental regulations, laws, rules and statutes/acts.
• Describe the importance of data generation, maintaining data lifecycle, data governance and data
• Reliability throughout the lifecycle of the document.
• The Quality Management System should include sufficient instructional detail to facilitate a common understanding of the requirements, in addition to providing for sufficient recording of the various processes and evaluation of any observations, so that ongoing application of the requirements may be demonstrated. Control Provision must be available for documentation practices
OBJECTIVE OF DOCUMENTATION SYSTEM
• Establish, Control, Monitor and record all activities, which directly or indirectly impact on all aspects of the quality of medicinal products.
• Appropriate good documentation practice should be applied with respect to the type of document
• Ensure that the Document should maintained Accuracy, Integrity, Availability and Legibility during the Document life Cycle.
• Document should be free from Error and during any point of if error identify then rectify with proper reason for correcting including sign and date.
• The Term “Written” in any document Means Recorded /Document on Media from which data may be rendered in a Human Readable form.
• Site Master File: A document describing the GMP related activities of the manufacturer.
Two primary types of documentation
a) Instructions (directions, requirements
Specifications, Manufacturing Formulae, processing, Packaging and testing Instructions Procedures, Protocols, Technical agreements
b) Records/reports.
Record, Certificate of Analysis, Reports
Definition:
Specifications - Describe in detail the requirements with which the products or Materials used or obtained during manufacture have to conform. They serve as a basis for quality evaluation.
Manufacturing Formulae, Processing, Packaging and Testing Instructions:
Provide detail all the starting materials, equipment and computerized systems (if any) to be used and specify all processing, packaging, sampling and testing instructions. In process Controls and process analytical technologies to be employed should be specified where relevant, together with acceptance criteria.
Procedures: (Otherwise known as Standard Operating Procedures, or SOPs), give Directions for performing certain operations.
Protocols: Give instructions for performing and recording certain discreet operations.
Technical Agreements: Are agreed between contract givers and acceptors for outsourced Activities.
Records: Provide evidence of various actions taken to demonstrate compliance with Instructions, e.g. activities, events, investigations, and in the case of manufactured Batches a history of each batch of product, including its distribution. Records include the raw data, which is used to generate other records. For electronic records regulated users should define which data are to be used as raw data. At least, all data on which quality decisions are based should be defined as raw data.
Certificates of Analysis: Provide a summary of testing results on samples of products or materials together with the evaluation for compliance to a stated Specification.
Reports: Document the conduct of particular exercises, projects or investigations, together with results, conclusions and recommendations.
ALCOA+: A commonly used acronym for ‘Attributable, Legible, Contemporaneous, Original and Accurate’ which puts additional emphasis on the attributes being ‘Complete, Consistent, Enduring and Available’– qualities which are implicit in the basic ALCOA principles.
Effective Date: It is the date of the document after which it becomes ready for actual use.
Error: A mistake in a document that is observed after a document was printed /executed.
GMP Documents: All types of documents which have direct or indirect impact on all aspects of the quality of drug substances/products and which are required to demonstrate or provide evidence of adherence to GMP standards and/or any other applicable regulatory requirements, are collectively referred as ‘GMP documents.’
Review: To look over, study or examine something with the aim of verifying the accuracy of the data.
Approved by/Certified by/Authorized by: Such a remark records the person who is responsible for approving GxP documents based on their evaluation of the conclusion(s).
EU GMP Chapter Points Summary
Table 1- 4.1 to 4.32 Points and these points can be sub-divided into various points
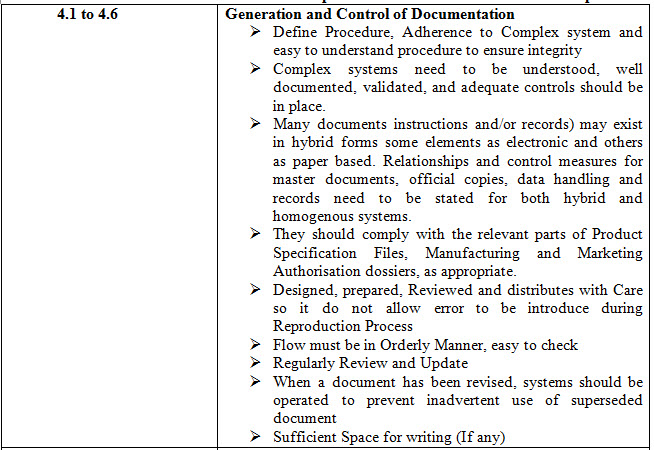
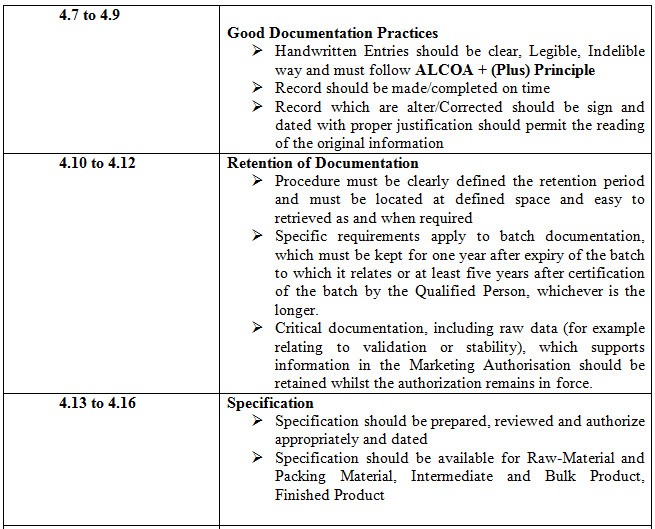

CONCLUSION
Based on review of EU documentation chapter and MHRA Presentation on Good Documentation Practices it is concluded that when an document is generated it must have provision for detail of person involve in preparing, checking, reviewing, authorizing before implementation and unique document number with revision provision, effective date, next review date and distributed with utmost care.
When a document has been revised, systems should be operated to prevent inadvertent use of superseded document and all GMP Document must be in Human Readable format either in printed or electronic form or photo-media form. Record which are alter/Corrected should be sign and dated with proper justification should permit the reading of the original information
A system should be in place to indicate special observations and any changes to critical data. GMP Document must have predefined retention period and document must be stored in secure and easy to retrieved or easily available as and when required.
Batch Processing and Packaging Instruction must be in place and Contemporaneous entry provision must be available. Instruction and procedure for using equipment; instrument must be clear and specific. Specification with authorization should be available for analysis of Raw Material and Packing Material, Intermediate and Bulk Product, Finished Product.
REFERENCES:
1. EudraLex Volume 4, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Chapter 4: Documentation Effective 30 June 2011.
2. MHRA Presentation - Good Documentation Practice a refresher Session 2, 2018.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








