
{ DOWNLOAD AS PDF }
 ABOUT AUTHORS
ABOUT AUTHORS
Surya Pratap Singh *, Kailash Dhaker, Abhisek Namdev, Mahaveer Prasad Khinchi, Surbhi Bhatnagar
1Department of pharmaceutics, Kota College of Pharmacy,
Kota, Rajasthan, India
sp.kota91@gmail.com
ABSTRACT
Process validation is the process for improving the safety and quality of the dosage form which is manufactured in the pharmaceutical industry. Basically, Process validation emphasize the role of objective measure and statistical tools and analyses knowledge ,detection ,and control of variability and give assurance on consistent of quality / productive throughout life cycle of product. Result from Process validation method can be used to judge the quality and consistency of analytical result. The purpose of this review to cover need of process validation, principle of process validation, type of process validation, phase of process validation, strategy for process validation. In this review article we discussed about the importance and strategy of validation of analytical procedure.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2451
|
PharmaTutor (ISSN: 2347 - 7881) Volume 4, Issue 12 Received On: 06/07/2016; Accepted On: 09/08/2016; Published On: 01/12/2016 How to cite this article: Singh SP, Dhaker K, Namdev A, Khinchi MP, Bhatnagar S; A Review on Process Validation; PharmaTutor; 2016; 4(12); 9-19 |
INTRODUCTION
Validation is an act of proving that procedure, process, equipment, material, activity or system perform as expected under given set of condition and also give the required accuracy, precision, sensitivity, ruggedness, etc. Method for process validation is the process used to confirm that the analytical procedure employed for a specific test is suitable for its intended use. Result from method validation can be used to judge the quality and consistency of analytical result it is an integral part of good analytical practice. The process is developed in such a way that the required parameter are achieved and it ensures that the output of the process will consistency meet required parameter during the routine production.
Purpose of validation
Validation is defined as “a documented program that provides a high degree of assurance that a specific process, method, or system will consistently produce a result meeting pre-determined acceptance criteria.”
The purpose of validation is to demonstrate the capability of the water treatment and air handling system to continuously supply the required quantity of water and air with the specified quality attributes. “Documented” means to provide documented “evidence.” Validation provides the system owner with the means of assessing when a water treatment and/or air handling system is operating outside established control parameter limits and provides a means for bringing the system back into a state of control. It results in written operating and maintenance procedures for personnel to follow, which in turn helps ensure consistent system performance.[1,2]
Method validation
Method validation is vast area which includes many validation parameters with different approaches for different level of requirement based on intended use of analytical method, criticality and regulatory requirements. Validated method also can give the unpredicted or unknown problem during the course of routine usage, because validated method has also limited level of confidence, as method was validated for known or predicted variable parameters or every method can fail sooner or later. But still after method development it needs to be validated as per requirement which gives certain level of confidence for its intended use.[3]
The type and degree of validation depends on the nature of the test. In particular, methods described in pharmacopeias may not have to be validated, but those should be verified. Different test methods require different validation parameters; as development of the project progresses and as analytical and product-specific information is acquired, the analytical methods evolve gradually updated. Each company has its own approach and set of acceptance criteria for different analytical assays, but these approaches must be within the confines of their line unit quality assurance department and be in accordance with any regulatory provisions. In this section, a description for each of the parameters to be validated (figures of merit) is described in details.[5]
Once an analytical method is developed for its intended use, it must be validated. The extent of validation evolves with the drug development phase. Usually, a limited validation is carried out to support an Investigational New Drug (IND) application and a more extensive validation for New Drug Application (NDA) and Marketing Authorization Application (MAA). Typical parameters recommended by FDA, USP, and ICH.[4]
1. Specificity
2. Linearity & Range
3. Precision
(i) Method precision (Repeatability)
(ii) Intermediate precision (Ruggedness)
4. Accuracy (Recovery)
5. Solution stability
6. Limit of Detection (LOD)
7. Limit of Quantification (LOQ)
8. Robustness
[adsense:468x15:2204050025]
Advantages of analytical method validation
- The advantages of the analytical method validation are as follow:
- The biggest advantage of method validation is that it builds a degree of confidence, not only for the developer but also to the user.
- Although the validation exercise may appear costly and time consuming, it results inexpensive, eliminates frustrating repetitions and leads to better time management in the end.
- Minor changes in the conditions such as reagent supplier or grade, analytical setup are unavoidable due to obvious reasons but the method validation absorbs the shock of such conditions and pays for more than invested on the process.[6,7]
IMPORTANCE OF VALIDATION
Planning for Validation
All validation activities should be planned. The key elements of a validation programme should be clearly defined and documented in a validation master plan (VMP) or equivalent documents.
The VMP should be a summary document, which is brief, concise and clear. The VMP should contain data on at least the following:
1. Validation policy.
2. Organizational structure of validation activities.
3. Summary of facilities, systems, equipment and processes to be validated.
4. Documentation format: The format to be used for protocols and reports.
5. Planning and scheduling.
6. Change control.
7. Reference to existing document.
8. In case of large projects, it may be necessary to create separate validation master plans.
Documentation
A written protocol should be established that specifies how qualification and validation will be conducted. The protocol should be reviewed and approved. The protocol should specify critical steps and acceptance criteria. A report that cross-references the qualification and /or validation protocol should be prepared, summarizing the results obtained, commenting on any deviations observed, and drawing the necessary conclusions, including recommending changes necessary to correct deficiencies.
Validation set up
To establish the desired attributes. These attributes include physical as well as chemical characteristics. In the case of parenterals, these absence of pyrogens, and freedom from visible particles. Acceptance specifications for the product should be established in order to attain uniformity and consistently the desired product attributes, and the specifications should be derived from testing and challenge of the system on sound statistical basis during the initial development and production phases and continuing through subsequent routine production.
The process and equipment should be selected to achieve the product specification. For example; design engineers; production and quality assurance people may all be involved. The process should be defined with a great deal of specificity and each step of the process should be challenged to determine its adequacy. These aspects are important in order to assure products of uniform quality, purity and performance.[8]
1. Assurance of quality.
2. Time bound.
3. Process optimization.
4. Reduction of quality cost.
5. Nominal mix-ups, and bottle necks.
6. Minimal batch failures, improved efficiently and productivity.
7. Reduction in rejections.
8. Increased output.
9. Avoidance of capital expenditures.
10. Fewer complaints about process related failures.
11. Reduced testing in process and in finished goods.
12. More rapid and reliable start-up of new equipments.
13. Easier scale-up form development work.
14. Easier maintenance of equipment.
15. Improved employee awareness of processes.
16. Government regulation (Compliance with validation requirements is necessary for obtaining Approval to manufacture and to introduce new product).
TYPE OF PROCESS VALIDATION
- Prospective Validation
- Concurrent Validation
- Retrospective Validation
- Process Re-Validation
Prospective Validation
- This validation usually carried out prior to distribution either of a new product or a product made under a revised manufacturing process. It is a preplanned scientific approach and includes the initial stages of formulation development, process development, setting of process sampling plans, designing of batch records, defining raw material specifications, completion of pilot runs, transfer of technology from scale-up batches to commercial size batches, listing major process is executed and environmental controls. In Prospective Validation, the validation protocol is executed before the process is put into commercial use. A series of experiment should be designed to determine the criticality of these factors.
- Each experiment should be planned and Documented fully in an authorized protocol. All equipment, production environment and the analytical testing methods to be used should have been fully validated. Master batch documents can be prepared only after the critical parameters of the process have been identified and machine settings, component specifications and environmental conditions have been determined. Using this defined process a series of batches should be produced. In theory, the number of process runs carried out and observations made should be sufficient to allow the normal extent of variation and trends to be established to provide sufficient data for evaluation. It is generally considered acceptable that three consecutive batches/runs within the finally agreed parameters, giving product of the desired quality would constitute a proper validation of the process. In practice, it may take some considerable time to accumulate these data.
- Prospective validation should include, but not be limited to the following:
- Short description of the process.[12]
- Summary of the critical processing steps to be investigated.
- List of the equipment/facilities to be used (including measuring , monitoring/recording equipment) together with its calibration status.
- Finished product specifications for release.
- List of analytical methods, as appropriate.
- Proposed in-process controls with acceptance criteria.
- Additional testing to be carried out, with acceptance criteria and analytical validation, as appropriate.
- Sampling plan.
- Methods for recording and evaluating results.
- Functions and responsibilities.
- Proposed timetable.
Concurrent Validation
- A process where current production batches are used to monitor processing parameters. It gives of the present batch being studied, and offers limited assurance regarding consistency of quality from batch to batch. Concurrent Validation may be the practical approach under certain circumstances. Examples of these may be when:
- A previous validated process is being transferred to a third party contract manufacturer or to another site.
- The product is a different strength of a previously validated product with the same ratio of active/inactive ingredients.
- The number of lots evaluated under the Retrospective Validation were not sufficient to obtain a high degree of assurance demonstrating that the process is fully under control.
- The number of batches produced are limited.
- Process with low production volume per batch and market demand.
Retrospective Validation
Conducted fir a product already being marked, and is based on extensive data accumulated over several lots and over time. Retrospective Validation may be used for older products which were not validated by the fabricator at the time that they were first marketed, and which are now to be validated to confirm to the requirements of division of the Regulation to be Food and Drugs Act.
This is achieved by the review of the historical manufacturing testing data to prove that the process has always remained in control. This type of validation of a process for a product already in distribution.[13]
Some of the essential elements for Retrospective Validation are:
- Batches manufactured for a defined period (minimum of 10 last consecutive batches).
- Number of lots released per year.
- Batch size/strength/manufacturer/year/period.
- Master manufacturing/packaging documents.
- Current specifications for active materials/finished products.
- List of process deviations, corrective actions and changes to manufacturing documents.
- Data for stability testing for several batches.
Change control
Written procedures should be in place to describe the actions to be taken if change is proposed to the starting material, product component, process equipment, process environment (or site), method of production or any other change that may affect product quality or reproducibility of the process. Change control procedure should ensure that sufficient support data are generated to demonstrate that the revised process will result in a product of the desired quality, consistent with the approved specifications.
Process Re-Validation:
- Required when there is a change in any of the critical process parameters, formulation, primary packaging components, raw material fabricator, major equipment or premises. Failure to meet product and process specifications in batches would also require process re-validation.
- Re-Validation becomes necessary in certain situations. The following are examples of
- some of the planned or unplanned changes that may require re-validation:
- Changes in raw materials (physical properties such as density, viscosity, particle
- distribution, and moisture, etc., that may affect the process or product).
- Changes in the source of active raw material manufacturer. Changes in packaging material (primary container/closure system).
- Changes in the process (e.g., mixing time, drying temperatures and batch size).
- Changes in the equipment (e.g. addition of automatic detection system).
- Changes of equipment which involve the replacement of equipment on a “like for like”
- basis would not normally require a revalidation except that this new equipment
- Changes in the plant/facility.
- Re-Validation becomes necessary in certain situations.
PRINCIPLE FOR PROCESS VALIDATION
Installation Qualification (IQ):
- It is establishing by objective evidence that all key aspects of the process equipment and ancillary system installation adhere to the manufacturer’s approved specification and that the recommendation of the
- supplier of the *equipment are suitably considered.
- IQ considerations are:
- Equipment design features (i.e. material of construction clean ability, etc.)
- Installation conditions (wiring, utility, functionality, etc.)
- Calibration, preventative maintenance, cleaning schedules.
- Safety features.
- Supplier documentation, prints, drawings and manuals.
- Software documented.
- Spare parts list.
- Environmental conditions (such as clean room requirements, temperature, and humidity
Operational Qualification (OQ):
- It is establishing by objective evidence process control limits and action levels which result in product that all predetermine requirements.
- OQ considerations include:
- Process control limits (time, temperature, pressure, line speed, setup conditions, etc.)
- Software parameters.
- Raw material specifications
- Process operating procedures.
- Material handling requirements.
- Process change control.
- Training.
- Short term stability and capability of the process, (latitude studies or control charts).
- Potential failure modes, action levels and worst-case conditions.
- The use of statistically valid techniques such as screening experiments to optimize the process can be used during this phase.
Performance Qualification (PQ):
- It is establishing by objective evidence that the process, under anticipated conditions, consistently produces a product which meets all predetermined requirements.
- PQ considerations include:
- Actual product and process parameters and procedures established in OQ.
- Acceptability of the product.
- Assurance of process capability as established in OQ.
- Process repeatability, long term process stability.[13]
STAGES OF PROCESS VALIDATION
Process Validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product.[15]
The following stages were performed in Process Validation-
Stage 1 – Process Design:
“Focusing exclusively on qualification efforts without also understanding the manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities. It covers all activities relating to product research and development, formulation, pilot batch studies, scale-up studies, transfer of technology to commercial scale batches, establishing stability conditions, storage and handling of in-process and finished dosage forms, equipment qualification, installation qualification, master production documents ,operational qualification, process capability. Also this is the stage in which the establishment of a strategy for process control is taking place using accumulation knowledge and understanding of the process.”
Stage 2 – Process Qualification:
During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing. It confirm that all established limits of the Critical Process Parameters are valid and that satisfactory products can be produced even under “worst case” conditions. GMP compliant procedures must be followed in this stage and successful completion of this stage is necessary before commercial distribution of a product.
There are two aspect of process qualification;
(a) Design of facilities and qualification of equipment and utilities
- Proper design of manufacturing facility is desired under 21 CFR part 211, subpart C, of
- the CGMP regulation on Buildings and Facilities.
- Activities performed to assure proper facility design and that the equipment and utilities are suitable for their intended use and perform properly.
(b) Process Performance qualification
- “Criteria and process performance indicators that allow for a science and risk-based decision about the ability of the process to consistently produce quality products”.
- Part of the planning for stage 2 involves defining performance criteria and deciding what data to collect when, how much data, and appropriate analysis of the data.
- Likely consist of planned comparisons and evaluations of some combination of process
- measures as well as in-process and trial product attributes.
- Manufacturer must scientifically determine suitable criteria and justify it.
- Objective measures, where possible.
- May be possible to leverage earlier study data if relevant to the commercial scale.
Stage 3 – Continued Process Verification:
On going assurance is gained during routine production that the process remains in a state of control. The validation maintenance stage requires frequent review of all process related documents, including validation audit reports to assure that there have been no changes, deviations, failures, modifications to the production process, and that all SOPs have been followed, including change control procedures. A successful validation program depends on the knowledge and understanding and the approach to control manufacturing processes. These include the source of variation, the limitation of the detection of the variation, and the attributes susceptible of the variation.[16]
![]()
Table 1 : Continued Process Verification
|
Stage
|
Intent |
Typical activities |
|
Process Design |
To define the commercial process on knowledge gained through development and scale up activities. |
A combination of product and process design (Quality by Design-QBD). Experiments to determine process parameters, variability and necessary control. Risk assessments Other activities required to define the commercial Process. Design or experiment testing Facility design. Equipment & utilities qualification. |
|
Process Qualification |
To confirm the process design as capable of reproducible commercial manufacturing. |
Performance qualification (PQ). Strong emphasis on the use of statistical analysis of process data to understand process consistency and Performance. |
|
Continued Process Verification |
To provide ongoing assurance that the process remains in a state of control during routine production through quality procedures through quality procedures and continuous improvement initiatives. |
Procedure lased data collection from every batch. Data trending and statistical analysis product review. Equipment and facility maintenance calibration. Management review and production staff feedback.
Improvement initiative through process experience. |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESPONSIBILITY &VALIDATION PROTOCOL
Responsibility department and their responsibility for Process Validation
The validation working party is convened to define progress, coordinate and ultimately, approved the entire effort, including all of the documentation generated.
The working party would usually include the following discipline members, preferably those with a good insight into the company’s operation.[16]
Table 2: Responsibility department and their responsibility for Process Validation
|
Department or Designee |
Responsibility |
|
3rd Level of Process Engineer |
Prepare and review the validation protocol. Ensue regarding the Title, Market, Batch Size, Report no, Batch details, Product details, Reference documents. |
|
2nd Level of Process Engineer |
Responsible for execution of process validation batch. Ensure that the information regarding reason for validation, product specification & acceptance criteria, measuring device used, batch fabrication details, in-process characteristics, validation data, results & conclusion. |
|
1st Level of Process Engineer |
Review validation protocol and clarify validation report. Also ensure that batches are executed as per the plan and approved protocol. Prepare periodic revalidation calendar. |
|
Validation |
Review validation protocol and certify validation report. Review periodic revalidation calendar. |
|
2nd level of Quality Assurance Manager |
Responsible for withdrawing sample as defined in the validation protocol. Review the protocol with respect sampling plans and procedure, and validation sample analysis results. Responsible for analyzing the samples as per defined specification/procedure details in the protocol and responsible for review and approval of validation protocol and certify validation report. |
|
Head (Engineering) |
Review the equipment and area in perfect working condition as required shall certify the above in validation protocol and validation report. |
|
Manager Operation |
Review and ensure that the information regarding batch details, product details, pack details of input material, equipment used, batch fabrication details, in-process characteristics, yield monitoring, result & conclusion. |
|
Authorized Regulatory Person |
Review the batch details, product details, pack details of input material with respect to the regulatory requirements and approved dossier in case of commercialized products in the validation protocol and certify the validation report. |
|
Head (Quality Assurance) |
Approve the validation protocol for implementation and certify the validation report. |
Validation protocol
Detailed protocol for performing validations are essential to ensure that the process is adequately validated. Process validation protocols should include the following elements:
- Scope of coverage of the validation study.
- Objectives Validation team membership, their qualifications and responsibilities.
- Type of validation: prospective, concurrent, retrospective, re-validation.
- Number and selection of batches to be on the validation study.
- A list of all equipment to be used; their normal and worst case operating parameters.
- Outcome of IQ, OQ for critical equipment.
- Requirements for calibration of all measuring devices.
- Critical process parameters and their respective tolerances.
- Process variables and attributes with probable risk and prevention shall be captured.
- Description of the processing steps: copy of the master documents for the product.
- Sampling points, stages of sampling, methods of sampling, sampling plans.
- Statistical tools to be used in the analysis of data.
- Training requirements for the processing operators.
- Validated test methods to be used in process testing and for the finished product.
- Specifications for raw and packaging materials and test methods.
- Forms and charts to be used for documenting results.
- Format for presentation of results, documenting conclusions and for approval of study result[16,17]
Process Validation Decision
The following model may be useful in determining whether or not a process should be Validated
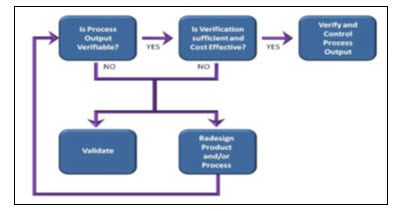
Process Validation Decision Tree
(i) Process Validation Decision Tree for change in process controls of manufacturing process of drug products:

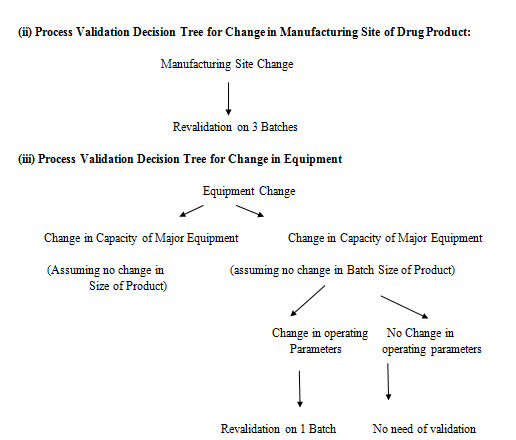
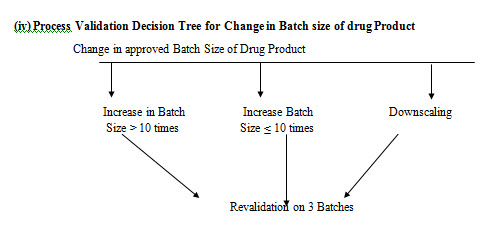
CONCLUSION
The pharmaceutical Process Validation is the most important and recognized parameters for in-process materials and finished product . The product should be designed robustly enough to withstand variations in the manufacturing process and the manufacturing process should be capable and stable to assure continued safe products that perform adequately. Process validation involves a series of activities taking place over the lifecycle of the product and process. Validation has been proven assurance for the process efficiency and sturdiness and it is the full fledged quality attributing tool for the pharmaceutical industries and making different type dosage form and solution. Validation is the commonest word in the areas of drug development, manufacturing and specification of finished products. It also renders reduction in the cost linked with process monitoring, sampling and testing. Apart from all the consistency and reliability of a validated process to produce a quality product is the very important for an industry.
The Importance of process validation are-Assurance of Quality-Validation is an extension of the concepts of quality assurance since close control of the process is necessary to assure product quality end product testing, in the absence of validation, gives little assurance of quality for variety reasons, among which are very limited sample size. The limited number of tests performed on a sample. For example, it is impractical to test for all potential impurities or contaminants. The limited sensitivity of the test.
Process Optimization-The optimization of a process for maximum efficiency, while maintaining quality standards, is a consequence of validation. Literal meaning of word to optimize is “To make as effective, perfect or useful as possible”. The optimization of the facility, equipment, systems, and processes results in a product that meets quality requirements at the lowest cost.
Reduction of quality costs -Finally it can be concluded that process validation is a key element in the quality assurance of pharmaceutical product as the end product testing is not sufficient to assure the quality of finished product
REFERENCES
- Sharma Ajay , Sharma Rohit “Validation of analytical procedure :a comparison of ICH Vs pharmacopeia” irjp.2012,3(6);39-42.
- Nash Robert A., Wachter Alfred H “pharmaceuticals processes Validation”. Drugs and the pharmaceutical sciences.1993, 29; 438.
- World Health Organization. Guidelines on the Validation of Manufacturing Processes.1993.
- M.J. Green, Anal. Chem’ Analytical Methods Validation’., 1996,68;305
- G.A. Shabir, J. Chromatogra. ;Analytical Methods Validation”.2003,987;57.
- Himanshu , pahuja Sonia “A review on pharmaceutical process validation “’international research journal of pharmacy” irjp 2012.
- Keyur b.ahir ,khushboo d.sing ,hetal s.patel “Overview of validation and basic concept of process validation”scholar academic journal of pharmacy 2014,3(2);178-190.
- Nandhakumar L., DharmamoorthyG., Rameshkumar S and Chandrasekaran S. “An Overview of Pharmaceutical Validation: Quality Assurance View Point” International journal of research in pharmacy and chemistry .2011,1(4);12.
- Ravichandran v ,shalini s, harish rajak “Validation analytical method strategies & importance”International journal of pharmacy and pharmaceutical science. 2010,2;120-139.
- Lower Stephen K. “Introduction pharmaceutical solution’ journal of pharmaceutical;2013;43;558-565.
- Frantsits Werner“Pharmaceutical solutions for oral administrationjournal of pharmaceutica.1999;39(12).
- Nandhakumar L., DharmamoorthyG., Rameshkumar S and Chandrasekaran S. “An Overview of Pharmaceutical Validation: Quality Assurance View Point” International journal of research in pharmacy and chemistry .2011,1(4);12.
- Md. Shoaib Alam Pharmaceutical Process Validation: An Overview. Journal of Advanced Pharmacy Education & Research. 2012,2(4);185-200.
- Nash R. A and Wachter A. H, “Pharmaceutical Process Validation an International” Drugs and the pharmaceutical sciences. 2003,3; 17 – 40.
- Kathiresan K*, Moorthi C, Prathyusha Y, Gade B. R, Reddy B. K, Manavalan R, ; An overview of pharmaceutical validation; Research Journal of Pharmaceutical, Biological and Chemical Sciences; ISSN: 0975-8585; October – December 2010; RJPBCS 1(4);1026.
- Md. Shoaib Alam Pharmaceutical Process Validation: An Overview. Journal of Advanced Pharmacy Education & Research. 2012, 2(4); 185-200.
- U.S. Food and Drug Administration. Guidelines on General Principles of Process Validation. 1987.
- Sanjay Bajaj, Dinesh Singla and Neha Sakhuja’ Stability Testing of Pharmaceutical Products’ Journal of Applied Pharmaceutical Science 02 (03); 2012: 129-138.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








