
{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
Gadhavi Aakash Vijaykumar*, Jaimin Kapadiya, Jitendra Patel, UM Upadhyay
Department of Pharmaceutics,
Sigma Institute of Pharmacy, Vadodara, Gujarat, India
*aakashgadhaviaks3377@gmail.com
ABSTRACT
The aim of the present work was to enhancement of Solubility and Dissolution of Clozapine using liquisolid technique. This work aimed to study the effect of different liquid vehicles on release characteristics of Clozapine. For liquisolid technique, different amount of liquid and ratio of carrier to coating material were employed. Avicel PH 102 and Aerosil 200 were used as carrier and coating material respectively and sodium starch glycolate (5%) was used as superdisintegrant. The empirical method as introduced by Spireas and Bolton 1999 was applied strictly to calculate amount of carrier and coating materials required to prepare liquisolid tablet. Quality control test like uniformity of tablet weight, uniformity of drug content, hardness, friability test, disintegration test and in-vitro dissolution tests were performed to evaluate the each batch of prepared tablets. In vitro drug dissolution profile of the liquisolid formulation were studied and compared with the Directly Compressed tablet of Clozapine in 0.1N HCL. Stability study was carried out to evaluate the stability of the tablet under humid condition. It was found to be that the optimized liquisolid tablet has high dissolution efficiency (80.44%) and lower mean dissolution time (MDT).From present study it can be concluded that liquisolid technique shows better improvement in solubility and dissolution of Clozapine than the pure drug.
INTRODUCTION
About 40% of the drug candidates identified via combinatorial screening programmers are poorly water soluble. The aqueous solubility for poorly water-soluble drugs is usually less than 100 µg/ml. The dissolution rate is the rate-limiting factor in drug absorption for class II (low solubility and high permeability) and class IV (low solubility and low permeability) drugs as defined in the Biopharmaceutics Classification System, BCS.[1-4]
Over the years, various techniques have been employed to enhance the dissolution profile and, in turn, the absorption efficiency and bioavailability of water insoluble drugs and/or liquid lipophilic medications. Various techniques like micronization, chemical modification, pH adjustment, solid dispersion, complexation, co-solvency, micellar solubilization, hydrotropy etc. The use of water-soluble salts and polymorphic forms, the formation of water-soluble molecular complexes, drug micronization, solid dispersion, co-precipitation, lyophilization, microencapsulation, and the inclusion of drug solutions or liquid drugs into soft gelatin capsules are some of the major formulation tools which have been shown to enhance the dissolution characteristics of water-insoluble drugs, however, among them, the technique of ‘‘liquisolid compacts” is one of the most promising techniques. [5-11]
The liquisolid technique as described by Spireas is a novel concept, where a liquid may be transformed into a free flowing, readily compressible and apparently dry powder by simple physical blending with selected carrier and coating material. The liquid portion, which can be a liquid drug, a drug suspension or a drug solution in suitable non-volatile liquid vehicles, is included into the porous carrier material. Inert, preferably water-miscible organic solvent systems with high boiling point such as liquid polyethylene glycols, propylene glycol, or glycerin are most excellent fitting as liquid vehicles. As the carrier is saturated with liquid, a liquid layer is formed on the particle surface which is instantly adsorbed by the fine coating particles. The liquisolid compacts are acceptably flowing and compressible powdered forms of liquid medications. The term ‘liquid medication’ refers to liquid lipophilic (oily) drugs or water-insoluble solid drugs dissolved in suitable water-miscible non-volatile solvent systems termed as the liquid vehicle. Such liquid medication may be converted into a dry-looking, nonadherent, free flowing and readily compressible powders by a simple admixture with selected powder excipients referred to as the “carrier and coating materials”. [12-17]
Spireas et al proposed the new mathematical model in accordance to retain good flow behavior and compressibility to design the formulation for Liquisolid technique. Mandatory requirements for this technique are suitable drug candidate, suitable non-volatile solvent, carrier and coating materials. The basic properties of powder are proposed according to Spireas et al is “Flowable liquid retention potential” (value) and compressible liquid retention potential” (ψ value). Flowable liquid retentional potential: defined as maximum weight of liquid (solvent) that can be retained per unit weight of powder (excipient) material to produce good flow. Compressible liquid retention potential: defined as the compression force applied to produce tablets with acceptable strength without squeezing out any liquid during compression. Excipient ratio (R): defined as Carrier to coating ratio quotated as
R= Q/q
Q= Carrier material,
q= Coating material.
Liquid load factor (Lf): defined as weight of liquid medicament (W) to weight of Carrier (w).
Lf = W/Q
The Ø value is for calculating excipients quantities. Equation is
Lf = Ø + Ø (1/R)
Where, Ø and Ø are values of carrier and coating material. [18-19]
Clozapine [8-chloro-11-(4-methylpiperazin-1-yl)-5Hdibenzo [b, e][1, 4]diazepine] is a potential antipsychotic agent and is also used for treatment of resistant schizophrenia. Clozapine has a yellow crystalline powder and melting point at 183°C and is slightly soluble in water. It is highly lipophilic and highly bound to plasma protein (97%). To achieve a high level of safety and effectiveness in pharmacotherapy, quality requirements of active substances are growing. The starting dose of CZ is 12.5 mg orally once, 25mg or twice a day. It is practically insoluble in water, having only < 27% - 50% oral bioavailability. CZ undergoes extensive first pass metabolism. Dosage adjustments may be needed based upon individual patient characteristics. CLZ functions by blocking dopamine receptors (D1, D2 to a lesser extent and D4) thereby countering schizophrenic effects. The use of clozapine is associated with side effects: extreme constipation, night- time drooling, muscle stiffness, sedation, tremors, orthostatic, hyperglycemia, and weight gain. The risks of extra pyramidal symptoms such as tardive dyskinesia are much less with clozapine when compared to the typical antipsychotics. Clozapine also carries eleven black box warnings for agranulocytosis, CNS depression, leukopenia, neutropenia, seizure disorder, bone marrow suppression, dementia, hypotension, myocarditis, orthostatic hypotension and seizures. To achieve maximum therapeutic effect with a low risk of adverse effects, controlled released preparations are preferred. The side effects could be lowered by controlling the drug release and by adjusting the absorption rate.[20-27]
Material and Methods:
Materials
Clozapine was provided by Chemdye Corporation, Rajkot, India. PEG 200, PEG 400, PEG 600, Tween 20, Tween 40, Tween 80, Propylene glycol (S.D. Fines, Mumbai) act as Non Volatile Solvent, Avicel 101, Avicel 102, Aerosil 200 (S.D.fines, Mumbai) act as Carrier and Coating Materials, and Sodium Starch Glycolate & Magnesium Stearate act as super disintegrant & Lubricant were used.
Methods
Saturation solubility studies [28-36]
The solubility of Clozapine in different non-volatile liquid vehicles that are commonly used for the formulation of liquisolid compacts, namely, propylene glycol (PG), polyethylene glycol 200 (PEG 200), and PEG 400, PEG 600, Tween 20, Tween 40 and Tween 80, was determined by preparation of saturated solutions of the drug in these solvents and measuring their drug concentration. Excess Clozapine was stirred in the above mentioned solvents for 48 h at 25ºC. Accurately weighed quantities of the filtered supernatants were further diluted with methanol and analyzed spectrophotometrically at 252 nm for their drug content. From these results, the solubility of Clozapine in the respective liquid vehicle was calculated. Each experiment was carried out in triplicate.
Determination of the Flowable Liquid Retention Potential for carrier and coating material[29, 30, 33, 35, 36]
In constant weight of carrier/ coating material, incresing amount of solvent was incorporated and on each addition angle of repose was determined. The flowable liquid-retention potential (Ø -value) of each liquid/powder admixture was calculated using the following equation.
Ø -value = weight of liquid/weight of solid
The Ø -values were plotted against the corresponding angle of repose (for optimal flow properties). Corresponding to 33o of a liquid/powder admixture represented the flowable liquid-retention potential.
Determination of liquid load factors (Lf)[29, 30, 33, 35, 36]
The appropriate amounts of carrier and coating materials to produce acceptable flowing and compactible powders were calculated using Eqs.
Lf= ØCA + ØCO (1/R)
Based on the physical properties of powders termed ‘‘flowable liquid-retention potential” (Ø-value). The maximum amount of liquid loads on the carrier material, termed ‘‘load factor” (Lf).
Flowability of Clozapine liquisolid powders[29, 30, 33, 35, 36]
The flowability evaluation of each formula was carried out by fixed height method. As a general guide, powders with angles of repose greater than 50° have unsatisfactory flow properties, whereas minimum angles close to 25° correspond to very good flow properties.
The tablets were also evaluated for other different parameters like weight variation, friability, hardness, disintegration time.
Preparation of Liquisolid compacts [29, 30, 33, 35, and 36]
The desired quantity of the previously weighed solid Clozapine was dissolved in liquid vehicle (PEG 400). The solution was then sonicated for 15 min until a homogeneous drug solution was obtained. Next, the calculated weights (W) of the resulting liquid medications (equivalent to 25 mg drug) were incorporated into the calculated quantities of the carrier Avicel PH 102 and mixed thoroughly. The resulting wet mixture was then blended with the calculated amount of the coating material Aerosil 200 using a standard mixing process to form simple admixture. Finally 5% w/w of sodium starch glycolate and 0.75% magnesium Stearate was mixed with the above mixture for 10 min. The final blend of liquisolid powder system was compressed into tablets of desired weight of 25 mg strength each by using 9 station tablet compression machine flat faced punch and die size of 12 mm were used. Directly compressed conventional tablets (CND) which is used for comparisons with liquisolid compacts is prepared by directly compressing powder mixture of Clozapine with Avicel PH 102, Aerosil 200, sodium starch glycolate and Magnesium Stearate.
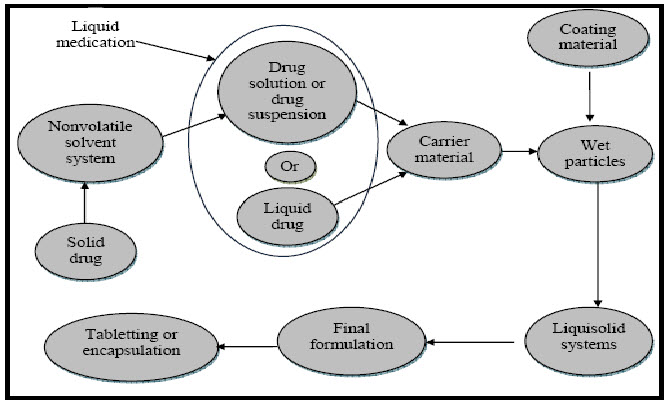
Fig. 1: Method of Preparation of Liquisolid Tablets
FORMULATION TABLE
Table 1: Formulation Composition of Liquisolid tablet
Dose of drug: 25 mg in each tablet
|
Formulation |
Amount of liquid (PEG 400) (mg) |
Ratio of carrier to coating material (R) |
Liquid load factor (Lf) |
Amount of Avicel PH 102 (mg) (Q = W/Lf) |
Aerosil 200 (mg) (q= Q/R) |
Mg. Stearate (mg) |
SSG (mg) |
Tablet Weight (mg) |
|
F1 |
250 |
5:1 |
0.448 |
558.03 |
111.6 |
7.08 |
47.20 |
998 |
|
F2 |
250 |
10:1 |
0.372 |
672.04 |
67.2 |
7.60 |
50.70 |
1071 |
|
F3 |
250 |
15:1 |
0.360 |
694.44 |
46.29 |
7.61 |
50.75 |
1072 |
|
F4 |
250 |
20:1 |
0.334 |
748.5 |
37.42 |
7.95 |
53.00 |
1121 |
|
F5 |
125 |
5:1 |
0.448 |
279.01 |
55.80 |
3.63 |
24.20 |
512 |
|
F6 |
125 |
10:1 |
0.372 |
336.02 |
33.60 |
3.89 |
25.95 |
548 |
|
F7 |
125 |
15:1 |
0.360 |
347.22 |
23.14 |
3.90 |
26.00 |
550 |
|
F8 |
125 |
20:1 |
0.334 |
374.25 |
18.71 |
4.06 |
27.10 |
548 |
|
F9 |
83 |
5:1 |
0.448 |
185.26 |
37.05 |
2.47 |
16.50 |
349 |
|
F10 |
83 |
10:1 |
0.372 |
223.11 |
22.31 |
2.64 |
17.65 |
374 |
|
F11 |
83 |
15:1 |
0.360 |
230.55 |
15.37 |
2.64 |
17.65 |
373 |
|
F12 |
83 |
20:1 |
0.334 |
248.50 |
12.42 |
2.76 |
18.40 |
390 |
|
F13 |
62 |
5:1 |
0.448 |
138.39 |
27.67 |
1.89 |
12.65 |
266 |
|
F14 |
62 |
10:1 |
0.372 |
166.66 |
16.66 |
2.02 |
13.51 |
285 |
|
F15 |
62 |
15:1 |
0.360 |
172.22 |
11.48 |
2.03 |
13.53 |
286 |
|
F16 |
62 |
20:1 |
0.334 |
185.62 |
9.28 |
2.11 |
14.09 |
295 |
All formulations contain 5% SSG and 0.75% magnesium stearate
Evaluation of Clozapine Liquisolid tablets [28, 29, 30, 33, 35, 36]
(Uniformity of tablet, drug content uniformity, tablet hardness, friability and disintegration tests):
For uniformity of tablet weight, 20 tablets were taken randomly from each tablet formulation and weighed individually. The average weight of all tablets and percentage deviation from the mean for each tablet were determined.
For uniformity of drug content, 10 tablets from each batch were taken randomly to examine its content uniformity. Each tablet was weighed and crushed individually. The crushed tablet powders were dissolved in methanol. The solution was filtered using glass microfiber filters (Whatman Filter Paper). The drug content was measured using UV/vis spectrophotometer(UV-1700, shimadzu Inc. Japan) at 252 nm. The percentages of individual drug content were calculated against the average drug content.
Hardness was measured using Monsanto hardness tester in terms of kg/cm2. Average hardness of three tablets was taken to study the reproducibility.
Six tablets from each batch were exposed to roche friability test apparatus for 100 rotations and percentage loss in weight was measured against initial weight.
%Friability (F) = {1-(W/W0)} x100
Where,
W0=Initial Weight of tablet
W=Weight of tablets after the test
The disintegration test was carried out using disintegration test apparatus USP (Hicon, India) using distilled water as disintegration medium. One tablet was introduced into each tube and a disc was added to each tube. Assembly was suspended in the beaker containing 900 ml distilled water. Time for disintegration of all six tablets was noted down.
In-Vitro Drug Release Study[28, 29, 30, 33, 35]
In vitro dissolution studies were performed on the prepared Clozapine liquisolid tablets using the USP dissolution apparatus II.Test was performed in 900ml of dissolution medium (0.1 N HCL). In all studies, the temperature of the dissolution medium was maintained at 37±0.5 °C and paddle speed at 50 rpm. The aliquots of 5ml were withdrawn at regular time intervals 10, 20, 30, 40, 50 and 60minutes, filtered, and analyzed spectrophotometrically at 243 nm.
For assessment and comparison, drug dissolution rates (DR) of drug were used. For this, amount of drug (in μg) dissolved per min that presented by each tablet formulation during the first 10 min were calculated as follows:
DR= (M ×D) / 1000
Where M is the total amount of Clozapine in each tablet (in this study, it is 25000 µg) and D denotes percentage of drug dissolved in first 10 min.
Dissolution Efficiency[35, 37]
Percentage dissolution efficiency (%DE) was calculated from the area under each dissolution curve at time “t” and measured using the trapezoidal rule and expressed as a percentage of the area of the rectangle described by 100% dissolution at same time was calculated.
The equation of %DE as follows:

Mean Dissolution Time (MDT)[35, 37]
Mean Dissolution Time is calculated from the following equation;
n
∑ tjΔMj
MDT = j=1
-------
n
∑ ΔMj
j=1
Where j is the sample number, n is the number of dissolution sample times, tj is the time at midpoint between tj and tj-1 and ΔMis the additional amount of drug dissolved between tj and tj-1.
Comparison of Dissolution Profile[35, 37]
For comparison between dissolution profiles of different samples, model independent mathematical approach of calculating a similarity factor f2 proposed by Moore and Flanner, 1996 was used. The similarity factor is a measure of similarity in the percentage dissolution between two dissolution curves and is defined by following equation:
where n is the number of withdrawal points, Rt is the percentage dissolved of reference at the time point t and Ttis the percentage dissolved of test at the time point t. A value of 100% for the similarity factor suggests that the test and reference profiles are identical. Values between 50 and 100 indicate that the dissolution profiles are similar whilst smaller values imply an increase in dissimilarity between release profiles.
The difference factor (f1) measures the percent error between two curves over all time points.
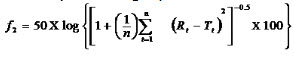
Where n is the sampling number, Rj and Tj are the percent dissolved of the reference and test products at each time point j. The percent error is zero when the test and drug reference profiles are identical and increase proportionally with the dissimilarity between the two dissolution profiles.
RESULT AND DISCUSSION:
Clozapine was selected as the model drug for these studies, since it is very slightly water soluble Drug and, thus, an ideal candidate for testing the potential of rapid release liquisolid compacts. In addition, The solubilities of Clozapine in propylene glycol, polyethylene glycol (PEG) 200, PEG 400, PEG 600, Tween 20, Tween 40, Tween 80 and Water determined in these studies, are given in Table 2.
Saturation Solubility Study
Table 2: Solubility of Clozapine in different solvents
|
Solvent |
Solubility (mg/ml) |
|
Water |
0.011 ±0.002 |
|
Tween 80 |
46.28 ±1.4 |
|
Tween 40 |
63.11 ±1.2 |
|
Tween 20 |
52.69 ±1.3 |
|
PEG 400 |
108.04 ±1.5 |
|
PEG 200 |
85.30±1.2 |
|
PEG 600 |
78.21±1.8 |
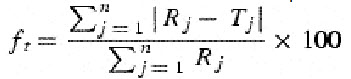
Figure 2: Solubility study graph of Clozapine different solvent
Clozapine shows highest solubility in PEG 400; it was selected for further studies and preparation of liquisolid tablets.
Flowable liquid retention potential (Ø-value) and liquid load factor (Lf)
Angle of repose was measured for powder containing Avicel PH101, Avicel PH102 and Aerosil 200. As Avicel PH 102 had shown higher liquid retention maintaining good flow than Avicel PH 101, it was used for preparation of liquisolid tablets. The Flowable Liquid retention potential (Ø-value) of Avicel 102 and Aerosil 200 shown in Table 3.
Table 3: Flowable liquid retention potential (Ø-value)
|
Ingredients |
Ø-Value |
|
Avicel PH 102 (CA) |
0.296 |
|
Aerosil 200 (CO) |
0.7624 |
Liquiload factor:
Lf= ØCA + ØCO (1/R)
For each R-value used, the corresponding Lf value can be calculated. As soon as the optimum liquid load factor Lf of a given excipients ratio is established for each formula and W is calculated according to Clozapine concentration in PEG400, the appropriate quantities of Avicel_ PH 102 (QCA) and Aerosil 200 (qCO) required to convert a given amount of liquid medication (W) into an acceptably flowing and compressible liquisolid system, were calculated in Table 1represents the exact qualitative and quantitative composition for each formula.
Table 4: Relationship between Ratio of carrier to coating material (R) and Liquiload factor (Lf)
|
Carrier to coating material ratio (R) |
Liquiload factor (Lf) |
|
5 |
0.448 |
|
10 |
0.372 |
|
15 |
0.360 |
|
20 |
0.334 |
Evaluation of Clozapine liquisolid tablets:
The collective data concerning Clozapine content in the tablet formulations, Angle of Repose, Thickness, tablets friability, hardness, disintegration times and Weight Variationare presented in Table 5.Thickness and Hardness were in the range of 2- 4.2 mm and 2-5.8 kg/cm2 respectively. In general, formulation should be directed at optimizing tablet hardness without applying excessive compression force, while at the same time assuring rapid tablet disintegration and drug dissolution. In other words, tablets should be sufficiently hard to resist breaking during normal
Handling and yet soft enough to disintegrate properly after Swallowing. The mean hardness of each liquisolid formula was determined and are presented in Table 5proving that all the liquisolid tablet formulation had acceptable hardness. The hydrogen bonds between hydrogen groups on adjacent cellulose molecules in Avicel PH 102 may account almost exclusively for the strength and cohesiveness of compacts, the high compressibility and compactness of Avicel PH 102 can be explained by the nature of the microcrystalline cellulose particles themselves which are held together by hydrogen bonds, when compressed, such particles are deformed plastically and a strong compact is formed due to the extremely large number of surfaces brought in contact during the plastic deformation and the strength of the hydrogen bonds formed. In addition, PEG 400 molecule contain two terminal hydroxyl groups, thus there is also a probability of forming hydrogen bonds with Avicel PH 102. All the Clozapine liquisolid tablets had acceptable friability as none of the tested formulation had percentage loss in tablets’ weights that exceed 1%; also, no tablet was cracked, split or broken in either formula. Since all the prepared formulation met the standard friability criteria, they are expected to show acceptable durability and withstand abrasion in handling, packaging and shipment and all the batches passed the wt. variation. Regarding disintegration time, it was found to be in the range of 12±9 to 118±12 min for liquisolid preparations intended for immediate drug release characteristics (Table 5), Faster disintegration time indicate rapid release rates. These are in accordance with dissolution rates (see below Figs. 3 and 4). Drug content was found to be 95.59 – 99.45%. All the parameters were within the range.
Table 5: Results of different tests on F1- F16
Table 5: Results of different tests on F1- F16
|
Formulation |
Angle of repose |
Thickness (mm) |
Hardness (kg/cm2) |
Friability |
DT (sec) |
% Content uniformity |
Weight Variation |
|
F1 |
33.0±0.5 |
4.0±0.2 |
3.7±0.2 |
0.38±0.03 |
90±10 |
97.45±0.747 |
Pass |
|
F2 |
37.5±0.2 |
2.9±0.3 |
4.9±0.1 |
0.43±0.02 |
72±12 |
95.59±0.82 |
Pass |
|
F3 |
32.3±0.3 |
3.2±0.2 |
3.3±0.2 |
0.55±0.06 |
88±11 |
98. 28±0.75 |
Pass |
|
F4 |
31.4±0.2 |
3.5±0.1 |
2.4±0.3 |
0.29±0.01 |
107±14 |
96.42±1.88 |
Pass |
|
F5 |
35.5±0.5 |
3.9±0.2 |
5.8±0.2 |
0.64±0.03 |
62±21 |
98.08±0.83 |
Pass |
|
F6 |
34.1±0.1 |
2.7±0.2 |
4.5±0.2 |
0.45±0.02 |
56±7 |
95.80±0.78 |
Pass |
|
F7 |
33.0±0.4 |
3.1±0.2 |
4.1±0.1 |
0.50±0.03 |
93±5 |
97.25±0.92 |
Pass |
|
F8 |
30.4±0.2 |
3.1±0.3 |
3.7±0.2 |
0.25±0.02 |
60±21 |
98.91±0.87 |
Pass |
|
F9 |
32.4±0.2 |
2.6±0.2 |
4.2±0.1 |
0.30±0.2 |
57±4 |
97.66±0.566 |
Pass |
|
F10 |
31.2±0.3 |
4.1±0.1 |
5.1±0.2 |
0.26±0.1 |
64±6 |
98.49±0.44 |
Pass |
|
F11 |
36.9±0.2 |
3.0±0.1 |
4.4±0.1 |
0.57±0.3 |
61±4 |
97.04±0.78 |
Pass |
|
F12 |
33.1±0.4 |
2.8±0.1 |
3.6±0.3 |
0.61±0.2 |
73±8 |
99.45±0.89 |
Pass |
|
F13 |
33.4±0.1 |
4.2±0.2 |
4.2±0.1 |
0.35±0.1 |
118±12 |
97.45±1.82 |
Pass |
|
F14 |
35.7±0.2 |
3.2±0.1 |
3.5±0.2 |
0.24±0.1 |
57±9 |
97.87±1.22 |
Pass |
|
F15 |
38.5±0.3 |
3.1±0.1 |
4.3±0.1 |
0.37±0.2 |
49±10 |
98.70±0.17 |
Pass |
|
F16 |
34.8±0.2 |
2.7±0.3 |
4.8±0.4 |
0.42±0.3 |
12±9 |
96.63±0.59 |
Pass |
(n=3), SD=standard deviation
RELEASE STUDY IN 0.1 N HCL:
The dissolution profiles of the selected Clozapine liquisolid tablet formulations together with the dissolution profile of Clozapine conventional, directly compressed tablets (DCT) are presented in Fig. 3. It was apparent that formula F8 has the highest dissolution pattern in both the rate and the extent of drug dissolved. The percentage of Clozapine dissolved from F8 reached 98.65% after only 50 min, while the directly compressed tablets had a maximum Clozapine content (85.02%) dissolved after 60 min. The percent of drug dissolved from each formula after 10 min (Q10) and the drug release rate (DR) were taken as a measure of the extent and the rate of drug dissolved from the prepared tablets, respectively. The results in the table clearly affirm that the liquisolid tablet formula F8 had the highest percentage of drug dissolved in 10 minutes; it dissolved 81.79% of its Clozapine content during the first 10 min.
Dissolution studies for the liquisolid formulations MDT and %DE of the liquisolid formulation
Calculated in media and reported in Table 6. DE values also increased with an increase in pH due to the high solubility of the drug at higher pH values. MDT values of the test product were low at all pH, indicating faster dissolution than Directly Compressed Tablet.
The increased dissolution of the liquisolid formulation is probably due to the drug presenting in a solubilised state in the formulation, which contributes to increased wetting properties, thereby enhancing the dissolute on rate. Similarly, the drug will be presented in a state of molecular dispersion as the formulation disintegrates in dissolution media. This will increase the effective
Surface area of the particles available for dissolution
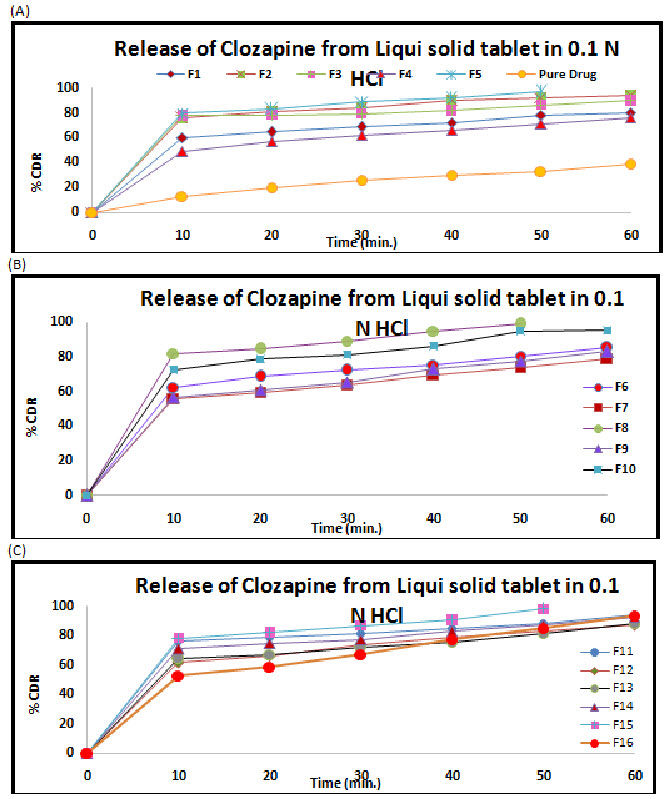
Figure 3: Release profile of Clozapine from liquisolid tablet in 0.1 N HCl (A) F1 to F5 and pure drug (B) F6- F10 and (C) F11- F16
The release profile of all formulations is shown in the Figure 3 respectively. F8 showed the maximum release 81.79 % to 98.65 % at 0.1 N HCL respective.
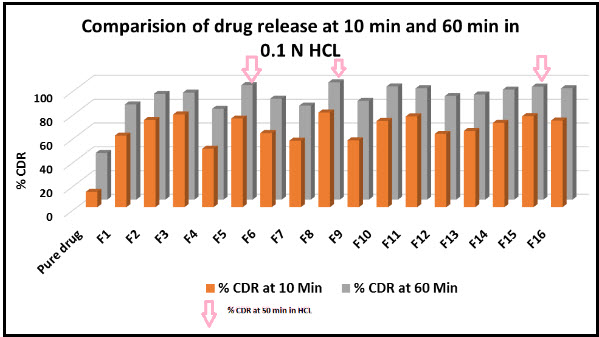
Figure 4: Comparison of the 10 min and 60 min dissolution rate of Clozapine exhibited by liquisolid tablets and pure drug.
Comparison with Directly Compressed Tablet
The release study of optimized liquisolid formulation (F8) was compared with the Directly Compressed Tablet of Clozapine. The release study was performed in 0.1N HCL. The release profile of both the formulation is shown in the Figure 5.
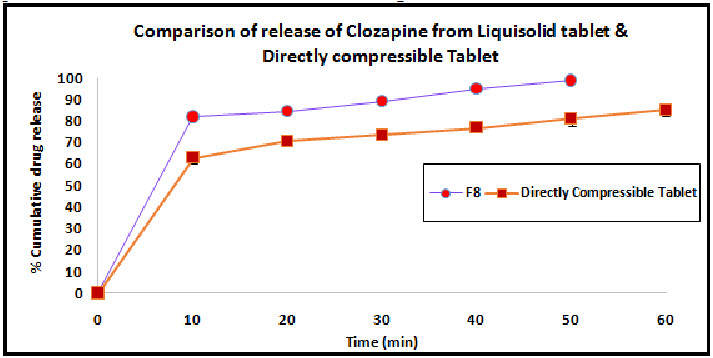
Figure 5: Comparison of liquisolid tablet and directly compressible tablet
Table 6: Dissolution Efficiency & Mean Dissolution Time
|
Batch |
Mean Dissolution Time |
%DE |
|
Pure Drug |
---- |
---- |
|
F1 |
12.20 |
79.66 |
|
F2 |
10.78 |
82.02 |
|
F3 |
10.14 |
80.41 |
|
F4 |
14.91 |
75.14 |
|
F5 |
9.61 |
80.77 |
|
F6 |
12.81 |
78.64 |
|
F7 |
14.27 |
76.21 |
|
F8 |
9.58 |
80.96 |
|
F9 |
14.85 |
75.11 |
|
F10 |
12.03 |
79.94 |
|
F11 |
13.75 |
66.05 |
|
F12 |
13.25 |
78.46 |
|
F13 |
14.25 |
76.22 |
|
F14 |
12.5 |
19.16 |
|
F15 |
11.61 |
80.64 |
|
F16 |
10.53 |
79.94 |
Similarity factor
The dissolution profile of the optimized batch (F8) & marketed formulation were compared to each other. The similarity factor (f2) was calculated. The f2 value was calculated to be 44.57 that indicate the dissimilarity between the two dissolution profiles of clozapine drug. The difference factor (f1) was measured for determination of percentage error between the two dissolution points. The f1 observed as 37.67 %, which indicates the dissimilarity between the two dissolution profiles.
Conclusion:
The aim of the present work was to enhance the solubility poorly water soluble drug like Clozapine using liquisolid technique. Clozapine is a BCS Class-II drug having low solubility and high permeability. Clozapine is insoluble in water (0.0118 mg/ml) and, as a consequence, it exhibits low and/or variable bioavailability after oral administration. For liquisolid technique, different amount of liquid and ratio of carrier to coating material were employed. Different evaluation tests like FT-IR, angle of repose, hardness, friability, disintegration time and in-vitro release were carried out.
Optimization of liquisolid tablets was done on the basis of dissolution characteristics and post compression parameters. The various dissolution parameters like dissolution rate in 10 min, dissolution efficiency were employed for the study. Results showed that liquisolid technique is a better technique for solubility enhancement of poorly soluble because there is no use of any organic solvent in the formulation.
The liquisolid tablet technique can prove to be an effective and efficient way for dissolution rate improvement of water insoluble drugs such as Clozapine as it shows faster release than that of pure drug. Polyethylene glycol 400 was used as a liquid vehicle. Enhanced dissolution rates obtained in the present study can be attributed to increased wetting and surface area available for dissolution. This novel approach to the formulation may be helpful to improve oral bioavailability. Thus it can be concluded that liquisolid technique is a potential way of solving solubility related problems encountered in pharmaceutical API.
References:
1. Tiong N, Elkordy A.A, ”Effect of liquisolid formulations on dissolution of naproxen” European Journal of Pharmaceutics and Biopharmaceutics, 2009, 73, 373–384.
2. C. Lipinski, Poor aqueous solubility – an industry wide problem in drug discovery, Am. Pharm. Rev. 5 (2002) 82–85.
3. D. Hörter, J.B. Dressman, Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract, Adv. Drug Deliv Rev. 46 (1997) 75–87.
4. R. Löbenberg, G.L. Amidon, Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards, Eur. J. Pharm. Biopharm. 50 (2000) 3–12.
5. Fahmy R.H, Kassem M.A, “Enhancement of femotidine dissolution rate through liquisolid tablets formulation: In vitro and In vivo evaluation” European Journal of Pharmaceutics and Biopharmaceutics, 2008, 69, 993–1003.
6. Vemula V.R, Legishetty V, Lingala S, “Solubility enhancement techniques – A Review” International Journal of Pharmaceutical Sciences Review and Research, 2010, 5(1), 41-47.
7. S. Spireas, M. Bolton, Liquisolid Systems and Methods of Preparing Same, U.S. Patent 5,968,550, 1999.
8. S. Spireas, Liquisolid Systems and Methods of Preparing Same, U.S. Patent 6,423,339 B1, 2002.
9. Sheth, C.I. Jarowski, Use of powdered solutions to improve the dissolution rate of polythiazide tablets, Int. J. Pharm. 16 (5) (1990) 769–777.
10. S. Spireas, T. Wang, R. Grover, Effect of powder substrate on the dissolution properties of methchrothiazide liquisolid compacts, Drug Dev. Ind. Pharm. 25 (2) (1999) 163–168.
11. A.Nokhodchi, The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts, J. Pharm. Pharma. Sci. 8 (1) (2005) 18–25.
12. Y. Javadzadeh,M.R. Siahi-Shadbad, M. Barzegar-Jalali, A. Nokhodchi, Enhancement of dissolution rate of piroxicam using liquisolid compacts, Il Farmaco 60 (2005) 361–365.
13. Syed I.H, Pavani E, “The liquisolid technique: Based Drug delivery system” International Journal of Pharmaceutical Sciences and Drug Research, 2012, 4(2), 88-96.
14. Spireas S. Liquisolid System and method of preparing same. U.S Patent 6423339B1, 2002.
15. Merisko E. Liversidge nanocrystals: resolving pharmaceutical formulation issues associated with poorly soluble compounds in: J.J matty (Ed), Particles, Marcel Dekker, Orlando, 2002.
16. Jarowski CI, Rohera BD, Spireas S. Powdered solution technology: principles and mechanism. Pharm Res. 1992; 9: 1351-1358.
17. Barzegar JM, Javadzadeh Y, Nokhodchi A, Siahi-Shadbad MR. Enhancement of dissolution rate of piroxicam using liquisolid compacts. II Farmaco. 2005; 60: 361-365.
18. Nokhodchi A, Hentzschel CM, Leopord CS. Drug release from liquisolid system: speed it up, slow it down. Expert Opin Drug Del. 2011; 8: 191-205.
19. Smirnova I, Suttiruengwong S, Seiler M, Arlt M. Dissolution rate enhancement by adsorption of poorly soluble drugs on hydrophilic silica aerogels. Pharm Dev Tech 2004; 9: 443-452.
20. Spireas S, Bolton M. Liquisolid Systems and Methods of Preparing Same. U.S. Patent 5,968,550, 1999.
21. Gowda D.V., Gupta V, Khan M and Bathool A, “Encapsulation of Clozapine in to Beeswax Microspheres: Preparation, Characterization and Release Kinetics” International Journal of PharmTech Research, 2011, (3, 4) 2199-2207.
22. USP DI, Drug information for the health care professional, 12th Ed. 974 -978, 1992.
23. Falkai P., Wobrock T., Lieberman J., Glenthoj B.,Gattaz W.F., Moller H.J & Wfsbp Task Force On Treatment Guidelines For Schizophrenia. The World Journal of Biological Psychiatry, 2006; 7(1): 5- 40.
24. Maxine X. Patel and Anthony S. David. Why aren't depot antipsychotics prescribed more often and what can be done about it? Advances in Psychiatric Treatment 2005; 11:203-211.
25. Kopparam Manjunath and Vobalaboina Venkateswarlu; Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration; Journal of Controlled Release Volume 107, Issue 2, 3 October 2005, Pages 215-228
26. http://www.drugs.com/search.php?searchterm=Clozapine
27. http://www.drugbank.ca/drugs/DB00363
28. http://www.medlineindia.com
29. Spireas S, Sadu S, “Enhancement of prednisolone dissolution properties using liquisolid compacts” International Journal of Pharmaceutics, 1998, 166, 177–188.
30. Elkordy A.A, Essa E.A, Dhuppad S, Jammigumpula P, “Liquisolid technique to enhance and to sustain griseofulvin dissolution: Effect of choice of non-volatile liquid vehicles” International Journal of Pharmaceutics, 2012, 434, 122–132.
31. Hentzschel C.M, Alnaief M, Smirnova I, Sakmann A, Leopold C.S, “ Enhancement of griseofulvin release from liquisolid compacts” European Journal of Pharmaceutics and Biopharmaceutics, 2012, 80, 130–135.
32. Javadzadeh Y, Musaalrezaei, Nokhodchi A,” Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices” International Journal of Pharmaceutics, 2008, 362, 102–108.
33. Javadzadeh Y, Siahi-Shadbad M.R, Barzegar-Jalali M, Nokhodchi A, “ Enhancement of dissolution rate of piroxicam using liquisolid compacts” II Farmaco,2005, 60, 361-365.
34. Shah C.V, Patel H.K., Shah V.H, Upadhyay U.M, “Design, Development and Optimization of Valsartan Liquisolid tablets using Box-Behnken Design” International Journal of Pharmaceutical Sciences and Research, 2012, 3(8), 2741-2753.
35. El-Say M. Khalid, Samy M. Ahmed, Fetouth I. Mohamed, “Formulation and evaluation of Rofecoxib liquisolid tablets” International Journal of Pharmaceutical Sciences Review and Research, 2010, 3(1), 135-142.
36. Naveen C, Shastri N, Tadikonda R.R, “ Use of the liquisolid compact technique for improvement of the dissolution rate of Valsartan ” Acta Pharmacutica Sinica B, 2012, 2 (5), 502-508.
37. Sathali A.A.H, C Deppa C, “ Formulation of liquisolid tablets of candesartan cilexetil ” International Journal of Research in Pharmaceutical Sciences, 2013, 4(2), 238-249.
REFERENCE ID: PHARMATUTOR-ART-2237
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 9 Received On: 30/06/2014; Accepted On: 02/07/2014; Published On: 01/09/2014How to cite this article: AV Gadhavi, J Kapadiya, J Patel, UM Upadhyay; Use of the Liquisolid Technique for Improvement of the solubility and Dissolution of Clozapine; PharmaTutor; 2014; 2(9); 101-114 |








