
{ DOWNLOAD AS PDF }
ABOUT AUTHORS
Nirav R.Soni, M.Pharm
A-one Pharmacy college,Enasan
Dept.of Qualtiy Assurance (QA)
nirav_sonic@yahoo.com
ABSTRACT
The specifications are to assure that each unit has the value of drug claimed on the label, that all the drug in each unit is out there for whole use ,that the drug steady within the formula in its certain final container for their expected shelf life and it’s having no toxic overseas substance. It’s greatly utilized in pharmaceutical enterprise and utilized by using wellness sector and support best which is finished via GMP, GLP and GCP and other organization including Pharmaceutical Quality System (PQS) , Quality Risk Management (QRM) and Quality by Design (QbD).
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2442
|
PharmaTutor (ISSN: 2347 - 7881) Volume 4, Issue 11 Received On: 03/06/2016; Accepted On: 21/06/2016; Published On: 01/11/2016 How to cite this article: Soni N; Specifications for Starting Materials, Intermediates and Finished Products; PharmaTutor; 2016; 4(11); 21-26 |
INTRODUCTION
Specifications for APIs and pharmaceutical drug products, both chemical components and biologics, are vital used for product quality and patient protection. The goals of specification arrangements are (1) to identify appropriate and safe limits or quantitative ranges during clinical development and (2) to give specifications for the product to enter the market. One of the most difficult challenges in establishing and consequently giving specifications is achieving the appropriate balance among all factors including human safety health and their efficacy, scientific proven data, analytical variability, process knowledge and capability, regulatory requirements, and business issues.[1] it is most important for a pharmaceutical company to have an effective and regulatory compliant approach to setting some requirements that can analyzed by both EMA and FDA requirements and expectations [2]. At each stage of drug development, from Phase 1 clinical trials via commercialization, the basic question needs to be addressed: ‘What is absolutely obligatory for the assigned specification to be doing well in getting the chemical drug/biologic components via clinical trials and into the worldwide market, but without impacting patient safety or creating delays in the programme?' of equal significant value is the require to describe which quality attributes do not need an assigned specification.[1,3] Regulatory obedient deficiencies in take off some specifications have resulted in clinical holds and market approval delays.
This course will support the attendee to establish specifications meeting global regulatory requirements and expectations. Participants may also come to be strong in justification of specifications.[4]
The term starting material has been absorbed where; regulatory change control and current good manufacturing practice are introduced into the synthesis of a various drug components. There is a drug components synthesis is shown in Figure 1. This conventional scheme depicts four regulatory steps and quite a lot of excellent manipulate points (specifications).
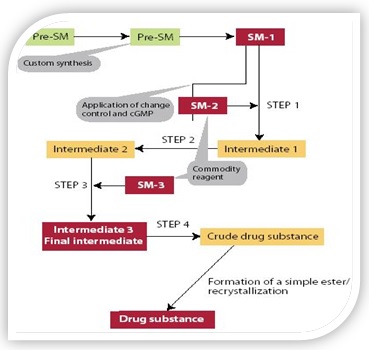
Figure 1: Schematic chart of regulatory drug substance synthesis.
Steps 1–4 includes a covalent bond formation. The various regulatory steps are disclosed in the Marketing Authorization Application and necessitate regulatory approval for changes. “Red Boxes” have the greatest regulatory importance. Materials in “bold text” are generally given a comprehensive and robust specification. “Orange Boxes” which are synthetic intermediates and can be isolated or remain in situ but are controlled using a more limited specification. (image source/getty images).[1,7,18]
Using a science and risk based framework, this article reviews the various regulatory guidelines in the EMEA, USFDA and MHLW. In addition, the authors address the International Conference on Harmonization (ICH) guidelines that recently impact the selection of starting materials for new drug components or substances for worldwide registration. The discussion takes in the initial publication with the introduction of various guidelines since ICH Q8 and Q9 “Pharmaceutical Development”, “QRM” respectively. and the withdrawal of FDA's BACPAC I and drug substance ICH guidance.[2-7,16]
[adsense:468x15:2204050025]
1.1 DEVELOPMENT OF SPECIFICATIONS:- Historical batch records of various pilot and production batches are usually the best source for establishment of meaningful specification and their development. Long run experience of production of same product always modifies specification required for finished product. A person authorized by Q.C. manager is responsible for approval of specifications. See below Table 1.[9-15]
Table 1: Criteria for Batch development.
|
Drug substance |
XYZ |
|
Proposed Potency limits |
93-107% |
|
Method |
Chloroform ext. followed by UV spectra |
|
Reproducibility |
+/-2% |
|
Batch Record |
99.3,98.4,103.4,97.9,101.3 98.9,101.8,99.7,98.6,100.4 |
|
Acceptable limits |
95-105% |
|
House limit |
96-104% |
1.1.1 FOLLOWING OBJECTIVES SHOULD BE COVERED IN DEVELOPMENT OF SPECIFICATION.[18-22]
[1].To ascertain which physical , chemical and biological characteristics of dosage form are critical which are helpful and which are not particularly important but are useful.
[2].To decide which dosage form characteristic shall be established as the criteria for evaluating routine production batches
[3].To establish the appropriate test methods for evaluating the selected criteria.
[4].To determine the acceptable tolerance and limit for each of the dosage form characteristic.
2. STARTING MATERIALS [23-29]:- Any substance of defined quality used in the production of pharmaceutical product, but excluding the packaging materials. Bulk Pharmaceutical Chemicals.
2.1. SPECIFICATIONS FOR STARTING MATERIALS [24-26]
(1)Generic and chemical name of the material.(2)Trade name or product code established by manufacturer.(3) Description (4) Name of pharmacopoeia (if official in pharmacopoeia)in which monograph appears or name of various recognized book of standards or international non-proprietary name(inn). (5) If not official in pharmacopoeia or any other book of standards, monograph to be employed for testing containing tests and limits for identity, their purity as well as physical and chemical characteristics microbiological standards (if any) and assay.(6)Approved suppliers.(7)Frequency of testing of stored material. (8) Special precautions to be taken during storage including safety aspects. (9) Date of issue of specification.
2.2 SPECIFICATIONS FOR THE DOCUMENTATION OF STARTING MATERIALS[23,27-29]
(1) Designated name and internal code reference(2)The reference if any to pharmacopoeial monograph.(3)Qualitative and quantitative requirements with acceptance limits.(4) Packaging material should decide to some specification and compatible with material or chemical substances.(5) Examined for defects and correctness of identity markings.(6) Documents should state necessary frequency for further assaying each starting material, as determined by its stability.(7) Specifications for starting materials are likely to identification, assay method or procedure , and other organic volatile impurities (limits for determined, undetermined, genotoxic and total identification ). In other cases, these requirements are most supplemented with those for extra residual solvents, catalysts or heavy metals and chirality.
3. SPECIFICATIONS FOR THE INTERMEDIATE:- Intermediate product is considered as one which is at any stage after dispensing of material and not got converted into packable bulk material. It should be accessible if these are bought or taken or if data obtained from intermediate products are used in the evaluation of the finished product. Some specification required like it should be kept under appropriate conditions. If purchased as such handled on receipt as though these are starting materials. [24]
4. SPECIFICATIONS FOR THE FINISHED PRODUCTS:- Finished products are those products which are ready for final dispatch to market or distribution. [23]
4.1.SPECIFICATIONS:- It should be some specification of two separate sets of manufacture (at release) and at the end of t90 A list of common characteristics, particular standards, tests and limits for the given results for the finished product must be provided. Another is All analytical test procedures used must be described in enough description like biological and microbiological methods where; relevant to enable the procedures to be repeated if necessary.[26,28]
The following control methods must be included in the specification[23-29]:- (1) General characteristics of the pharmaceutical form like physicochemical properties
(2) Identification tests of the active substances;
(3) Quantitative identification of active substances.
(4) Unlike there is appropriate justification, the maximum acceptable deviation in the active ingredients content of the finished product shall not exceed +/-5% at the time of manufacture.
(5) Purity tests (breakable products, residual solvents or extra process related impurities, microbial contamination).
(6) Pharmaceutical tests, e.g. dissolution;
(7) The identification tests for coloring materials used and identification and assay of antimicrobial or some chemical preservatives with beneficial limits. The preservatives content limits of 90-110 percent at release are suitable without further justification except in special cases.
(8) All procedures need to be validated. Results of the validation, comments on the in-house tests and standards must be provided.
(9) If the final product is tested on the basis of a monograph procedure given in a pharmacopoeia, it is ample to provide a copy of the monograph together with any test methods referenced but it is not copied in the standard monograph provide details of any specifications additional to those in the pharmacopoeia. It provides the results of validation of the assay procedure or method for this formulation.
(10) For pharmacopoeial methods, provide data which shows that the method is applicable to this formulation. Results of batch analysis (including the date and place of manufacture, batch size and use of batch tested) must be presented. The batch analysis must involve the results seen for all specifications at release.
4.2. SPECIFICATIONS FOR DOCUMENTATION OF FINISHED PRODUCTS which Include; (a) reference code and name (b)Names of actives (e.g. INN) (c)Formula (d) Dosage form, package details (e) Reference to sampling (f)Qualitative and quantitative requirements and limits (g) Storage conditions and precautions (h) Shelf life
4.3.SPECIFICATIONS FOR FINISHED PRODUCTS ALSO INCLUDE:-(a) Generic name of product (b)Trade name if any Dosage form and strength (c)Description including particulars such as colour, shape, dimensions , taste etc.(d) Physical properties such as weights or volumes(including limits) pH, viscosity, density, hardness, friability, disintegration time, dissolution time etc.(e)Name of pharmacopoeia or any other recognised book of standards in which its monograph appears, if not official, monograph to be employed for testing containing test for identity and purity, microbiological standards, if any biological tests, if any and assay; (f) Date of expiry. (g) Precautions during storage including safety aspects (h) Date of issue of specifications.
5. IMPORTANT SPECIFICATIONS FOR VARIOUS DOSAGE FORM[25-29]
5.1.TABLET &CAPSULE:- A)Physical appearance:- Size and Shape Identification marks, Thickness, Smell, Color, Shining, Texture B)Physical Tests are including various tests like Hardness , Disintegration test, Dissolution, Friability and Weight variation test C)Identification, assay, related substance tests D) Shelf-life determination(specially in new products)packaging & labeling I ) Outer Carton including Striping, Gum patta ,Finishing and Printing requirements II ) small pack(contains no. of strips) notifying Color & design, Printing matter, Finishing, Literature , Quantity of strips Iii)Strip Pack/Blister Pack involving leak test, printing matter and finishing IV)Tin Pack/Bottle Pack including Quality of tin/glass/plastic ,Printing matter , Quality of tablets/capsules, Lower & upper side cotton , Silica gel bag, Inserts are properly managed ,Outer carton packing
5.2. OINTMENT/CREAM :- A)Physical Appearance including Color, Texture ,Odor B)Physical Test including pH, Quantity determination, Foreign particle, Spreadability, Permeability, C) Identification, Assay, Related Substance Tests, D)Expiry Determination E)Packaging & Labeling E1) Outer Carton containing Striping, Gum patta, Finishing, Printing requirements E2)Small Pack including Color & design ,Printing matter, Finishing , Literature. Quantity of tubes E3) Tube involving Size of tube, color & shade, Leakage, Printing matter
5.3.LIQUID PREPARATION:- A)Physical Appearance including Color and odor of liquid and Visual inspection B)Physical Tests including Taste of liquid , pH ,Volume, PSD/GSD (suspension), Type of emulsion, SVR(suspension), Re-dispersibility and Test of creaming(emulsion) C) Identification, assay, related substance tests D) expiry determination E)Packaging & Labeling :-E1)Outer Carton :-Striping, Gum patta, Finishing and Printing requirements E2) Small Pack which includes Color & design, Printing matter, Finishing, Literature , Quantity of bottles E3)Tube notifying Size & shape of bottles ,Leakage , Cap quality , Color of glass, Printing matter
5.4. PARANTRAL PREPARATION
A)PHYSICAL APPEARANCE:-Color & clarity of liquid B)physical tests :- pH ,Volume, Leak test, Syringibility C)Identification, Assay, Related Substance Tests D)Special Requirements like Pyrogen testing and Sterility testing E)EXPIERY DETERMINATION F)PACKAGING & LABELLING F1)Outer Carton containing Striping, Gum patta, Finishing, Printing requirements F2)Small Pack containing Color & design, Printing matter, Finishing, Literature ,Quantity of vials/ampoules, Dropper in case of eye drops(quality & color of dropper) F3)Vials which containing Sealing check, Rubber stopper, Color specification of rubber stopper, Color specification of seal, Quality of glass, Color of glass vials and Diameter of vials F4)Ampoules involving Sealing, Quality of glass ,Color of glass ampoules, Diameter of ampoules, Uniform height and Leak-test.
5.5 SPECIFICATIONS FOR STORAGE OF PHARMACEUTICAL FINISHED PRODUCT [27-29]
(1) Finished product should be held id in quarantine until their final release, after which they should not be stored as unusable stock under conditions established by the manufacturer. (2) A separate area should be provided for the storage of the finished product. (3) Finished product may be stored according to their dosage form. (4)These storage areas should have adequate height and ventilation, adequate storage equipment like plant forms plastic and wooden , racks etc. nearby, to finished product area, an assembling and packaging area may be provided to execute sales orders, if space permits.
CONCLUSION
In the conclusion, every dosage forms having particular identity and test should be performed by analytical test including physical ,chemical and biological for some standard and specification. It covers some criteria and give quality of product not only but enhances the quality and characteristics for earlier stage to finished product even packaging. It has required for globalization and preparation of various novel dosage forms.
REFERENCES
1.John Geigert, The Center for Professional Innovation and Education (CfPIE) [Online] Available from: URL: https://www.cfpie.com/ProductDetails.aspx?ProductID=204
2.R.J. Timko et al., "Drug Substance Starting Materials: A Regulatory Perspective on Requirements and Selection," poster presented at American Association of Pharmaceutical Scientists Annual Meeting, Nashville, TN, [cited 2005 Nov].
3.ICH Q8 Pharmaceutical Development ; Geneva, Switzerland; [cited 2006 May].
4.ICH Q9 Quality Risk Management (Geneva, Switzerland, [cited 2006 June ].
5.FDA, Guidance for Industry: BACPAC I: Intermediates in Drug Substance Synthesis; Bulk Actives Post approval Changes: Chemistry, Manufacturing, and Controls Documentation, Feb. 2001, withdrawn, Fed. Regist. Notice [cited 2006 June 1].
6.FDA, Guidance for Industry: Drug Substance: Chemistry, Manufacturing, and Controls Information, Jan. 2004, withdrawn Fed. Regist. Notice [cited 2006 June 1].
7.ICH Q7 Good Manufacturing Guide For Active Pharmaceutical Ingredients, Geneva, Switzerland, [cited 2001 August].
8.FDA, Guidance for Industry: Guidance for Submitting Supporting Documentation in Drug Applications for the Manufacture of Drug Substances ; Rockville, MD, [cited 1987 Feb].
9.FDA, Guidance for Industry: Changes to an Approved NDA or ANDA, Rev. 1 Rockville, MD, [cited 2004 Apr].
10.EMEA Committee for Proprietary Medicinal Products, Guidance on the Chemistry of New Active Substances, CPMP/QWP/130/96, Rev 1 ; London, England, [cited 2003 Dec 17].
11.EU Guidelines to Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use, Part II, Basic Requirements for Active Substances used as Starting Materials (Brussels, Belgium, Oct. 2005).
12. EDQM Division Certification of Substances, Public Document PA/PH/Exp. CEP/T (06) 35, "Certification of Suitability of Monographs of the European Pharmacopoeia. How Can the Content of the Applications for a Certificate of Suitability for Chemical Purity Be Improved? The Top 10 Deficiencies found in applications"; Strasbourg, France [cited 2006 Dec].
13.MHLW, Pharmaceutical and Food Safety Bureau, Guidelines on Mentions in Manufacturing / Marketing Approval Application Dossiers for Pharmaceuticals and Others Based on Revised Pharmaceutical Affairs Law, PFSB/ELD 020001 Tokyo, Japan, [cited 2005 Feb 10].
14.FDA, Pharmaceutical CGMPs for the 21st Century: A Risk-Based Approach, Final Report; Rockville, MD [cited 2004 Sep].
15.ICH Q3A R2 Impurities in New Drug Substances Geneva,Switzerland [cited 2006 June ].
16.ICH Q3C Impurities: Residual Solvents (Geneva, Switzerland, Dec. 1997, and ICH Q3C Tables and Lists, Rev. [cited 2005 Nov 3].
17.ICH Q6A Specifications: Test Procedures and Acceptance Criteria For New Drug Substances and New Drug Products: Chemical Substances Geneva, Switzerland [cited 1999 Oct].
18.L. Muller et al., "A Rationale for Determining, Testing, and Controlling Specific Impurities in Pharmaceuticals That Possess Genotoxicity, "Regulatory Toxicology & Pharmacology 44:198–211, 2006.
19.EMEA Committee for Medicinal Products for Human Use, Guideline on the Limits of Genotoxic Impurities, CPMP/SWP/5199 London, England, [cited 2006 June 28].
20.Code of Federal Regulations, Title 21, Food and Drugs, Volume 3, Chapter 1, Subpart B: Food Additive Safety, Section 170.39: Threshold of Regulation for Substances Used in Food-Contact Articles ; General Services Administration, Revised [cited 2007 Apr 1].
21.D. Jacobson-Kram and T. McGovern, 'Toxicological Overview of Impurities in Pharmaceutical Products," Advanced Drug Delivery Reviews 59 (1) :38–42, 2007.
22.Potdar Manohar A. “Pharmaceutical quality assurance through CGMP , documentation and validation”, Nirali prakashan , (1):355-360
23.P.P.Sharma,” How to practice GMPs”, vandana publication (6):58-62
24.Essential Medicines and Health Products Information Portal.Available from: URL: http://apps.who.int/medicinedocs/en/cl/CL1.1/clmd,50.html
25.Kenneth E. Avis, Herbert A. Lieberman, and Leon Lachman , Marcel Dekker INC “Pharmaceutical dosage forms: Parentral medications”, 3(2) :58-62 .
26.Herbert A. Lieberman, Leon Lachman and Joseph B. Schwartz , Marcel Dekker INC “Pharmaceutical dosage forms: Tablets”,1(2):548-551.
27.Herbert A. Lieberman, Martin M.Rienger, and Gilbert Banker Marcel Dekker INC “Pharmaceutical dosage forms: Disperse system”, 3(2), 457-465.
28.National Pharmaceutical Control Bureau, Ministry of Health Malaysia :1-20
Available from :URL:
http://bpfk.moh.gov.my/images/Guidelines_Central/Guidelines_on_Veterinary/Guidelines-on-GMP-for-Veterinary-Premixes-January-2015.pdf
29.Graham T. Illing, Linda Billett, Robert J. Timko, “Drug Substance Starting Material Selection Pharmaceutical Technology”, 32( 12) , [cited 2008 Dec 2]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








