

{ DOWNLOAD AS PDF }
ABOUT AUHTORS
Sunil Shastri*, Harsh Narang.
Seth G.L. Bihani S.D. College of Technical Education,
Sriganganagar, Rajasthan.
sunil11044@gmail.com
ABSTRACT
The Combinatorial Chemistry is a scientific method in which a very large number of chemical entities are synthesized by condensing a small number of chemical compounds together in all combinations defined by a small set of chemical reactions.
Combinatorial technologies provided a possibility to produce new compounds in practically unlimited number. New strategies and technologies have also been developed that made possible to screen very large number of compounds and to identify useful components of mixtures containing millions of different substances. Instead of preparing and examining a single compound, families of new substances are synthesized and screened. In addition, combinatorial thinking and practice proved to be useful in areas outside the pharmaceutical research Such as search for more effective catalysts and materials research. Combinatorial chemistry became an accepted new branch within chemistry.
The aim of this project is to provide a basic introduction to the field of combinatorial chemistry describing the development of major techniques and some applications.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2491
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 5, Issue 5 Received On: 13/01/2017; Accepted On: 13/02/2017; Published On: 01/05/2017 How to cite this article:Shastri S, Narang H;Combinatorial Chemistry - Modern Synthesis Approach; PharmaTutor; 2017; 5(5);37-63 |
INTRODUCTION
Combinatorial chemistry is a collection of techniques which allow for the synthesis of multiple compounds at the same time. [Leard L. & Hendry A. (2007)]13
This nascent technology already produced more new compounds in just a few years than the pharmaceutical industry did in its entire history. Combinatorial chemistry has turned traditional chemistry upside down. It required chemists not to think in terms of synthesizing single, well-characterized compounds but in terms of simultaneously synthesizing large populations of compounds. [Miertus S. et al. (2000)]14
Combinatorial chemistry is one of the important new methodologies developed by researchers in the pharmaceutical industry to reduce the time and costs associated with producing effective and competitive new drugs.
By accelerating the process of chemical synthesis, this method is having a profound effect on all branches of chemistry, but especially on drug discovery. Through the rapidly evolving technology of combinatorial chemistry, it is now possible to produce compound libraries to screen for novel bioactivities. This powerful new technology has begun to help pharmaceutical companies to find new drug candidates quickly, save significant money in preclinical development costs and ultimately change their fundamental approach to drug discovery. [combichemistry.com]2
This branch of chemistry is very young, but in this short time it has had profound effects. This can be seen by its impact on medicinal chemistry and, in particular, the drug design process. Traditionally, potential lead compounds were synthesized one at a time. The biological activity of this compound was assayed, and the results would be reflected in the next round of design. This traditional method was useful, but time consuming and expensive. Computational chemistry led to more rational design of compounds to be tested, and high throughput screening led to quick in vitro assays. Synthesis of one compound at a time could no longer keep up, and thus became the rate limiting step in the process. Combinatorial chemistry was the solution to this problem. In combinatorial approach, one can cover many combinations An x Bn in one reaction Instead of doing multiple A x B type reactions.
Conventional Reaction: A + B------------------->AB
Combinatorial Chemistry: A1- n + B1- n--------->A1- n B1- n
Hence, In combinatorial chemistry, large numbers of compounds are made at the same time in small amounts, forming libraries which can be assayed for desired properties all at once. Finally the active compound is identified and made in quantity as a single compound. [Leard L. & Hendry A. (2007)]13
COMBINATORIAL CHEMISTRY APPROACH
Combinatorial chemistry may be defined as the systematic and repetitive, covalent connection of a set of different “building blocks” of varying structures to each other to yield a large array of diverse molecular entities. [Pandeya S.N. and Thakkar D. (2004)]16
Combinatorial chemistry encompasses many strategies and processes for the rapid synthesis of large, organized collections of compounds called libraries. The collection is then tested for the biological activity. Finally the active compound is identified and made in quantity as a single compound. [Lather V. et al. (2005)]12
Thus the combinatorial chemistry approach has two phases:
1. Making a library.
2. Finding the active compound. Screening mixtures for biological activity has been compared to finding a needle in a haystack. [Patel R. (2008)]17
In the past, chemists have traditionally made one compound at a time. For example compound ‘A’ would have been reacted with compound ‘B’ to give product ‘AB’, which would have been isolated after reaction work up and purification through crystallization, distillation or chromatography.
In contrast to conventional approach, combinatorial chemistry offers the potential to make every combination of compound ‘A1’ to ‘An’ with compound ‘B1’ to ‘Bn’
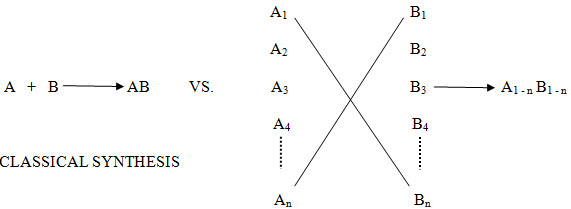
Figure -1. Comparison of Tradition and Combinatorial Strategies in Drug Design
The range of combinatorial techniques is highly diverse, and these products could be made individually in a parallel or in mixtures, using either solution or solid phase techniques. Whatever the technique used the common denominator is that productivity has been amplified beyond the levels that have been routine for the last hundred years. [combichemistry.com/principle.html]18
Combinatorial chemistry-a technology for creating molecules en masse and testing them rapidly for desirable properties-continues to branch out rapidly. Compared with conventional one-molecule-at-a-time discovery strategies, many researchers see combinatorial chemistry as a better way to discover new drugs, catalysts and materials. [Borman S. (1998)]1
The development of new processes for the generation of collection of structurally related compounds (libraries) with the introduction of combinatorial approaches has revitalized random screening as a paradigm for drug discovery and has raised enormous excitement about the possibility of finding new and valuable drugs in short times and at reasonable costs.
STRATEGIES
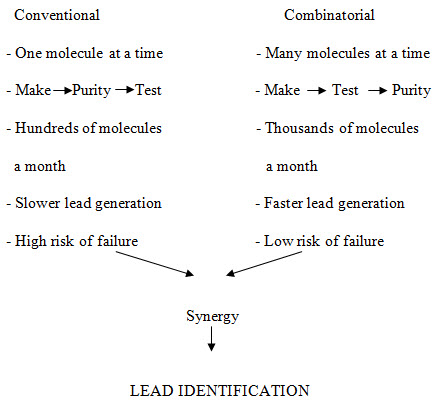
Figure -2. Principle characteristics of conventional vs. combinatorial strategy of drug discovery [Miertus S. et al. (2000)]14
PROS & CONES OF COMBINATORIAL CHEMISTRY
ADVANTAGES
(1) The creation of large libraries of molecules in a short time is the main advantage of combinatorial chemistry over traditional.
(2)Compounds that cannot be synthesized using traditional methods of medicinal chemistry can be synthesized using combinatorial techniques.
(3) Thecost of combinatorial chemistry library generation and analysis of said library is very high, but when considered on a per compound basis the price is significantly lower when compared to the cost of individual synthesis.
(4) More opportunities to generate lead compounds.
(5)Combinatorial chemistry speeds up drug discovery.
DISADVANTAGES
(1) Though combinatorial chemistry would solve all the problems associated with drug discovery, one still needs to synthesize the right compound.
(2) There is a limit to the chemistry you can do when using solid phase synthesis. The resin you use is often affected by the reaction types available and care must be taken so that the attachment of the reagent to the substrate and bead are unaffected. Each reaction step has to be carefully planned, and often a reaction isn't available because the chemistry affects the resin.
(3) While a large number of compounds are created, the libraries created are often not focused enough to generate a sufficient number of hits (Library components whose activity exceeds a predefined, statistically relevant threshold) during an assay for biological activity. There is a great deal of diversity created, but not often a central synthetic idea in the libraries. One can argue that there should be a focus on the type of molecule developed in order to maximize hits.
[Leard L. & Hendry A. (2007)]13
HISTORICAL DEVELOPMENT
Combinatorial chemistry is a very young science, having only been around for approxi-mately 20 years. It has been applied to drug design for an even shorter period of time. [Leard L. & Hendry A. (2007)]13
The origins of combinatorial chemistry can be traced back at least as far as 1963, when biochemistry professor R. Bruce Merrifield of Rockefeller University, New York City, developed a way to make peptides by solid-phase synthesis. [Borman S. (1998)]1
For his work on solid-phase synthesis, Bruce Merrifield won the Nobel Prize in chemistry in 1984. During this time, automated peptide synthesizer technology was in its infancy, and the preparation of individual peptides was a challenge. [Kim S. (2005a)]9
The field in its modern dimensions only began to take shape in the 1980s, when in 1984 research scientist H. Mario Geysen, now at Glaxo Wellcome, Research Triangle Park, N.C., developed a technique to synthesize arrays of peptides on pin-shaped solid supports in and in 1985 Richard Houghten developed a technique for creating peptide libraries in tiny mesh "tea bags" by solid-phase parallel synthesis. [Borman S. (1998)]1
Another early pioneer was Dr. Árpád Furka who introduced the commonly used split-and-pool method in 1988, which is used to prepare millions of new peptides in only a couple of days and also for synthesizing organic libraries. [Kim S. (2005a)]9
Through the 80’s and into the early 1990’s, combinatorial chemistry was focused on peptide synthesis and later oligonucleotide synthesis. [Leard L. & Hendry A. (2007)]13
In the 1990s, the focus of the field changed predominantly to the synthesis of small, drug like Organic com-pounds and many pharmaceutical companies and biotechnology firms now use it in their drug discovery efforts. [Mishra A.K. et al. (2010)]17
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
The major milestones in combinatorial chemistry can be represented in the following table:
|
Years |
Milestone |
|---|---|
|
1984 |
Limited peptide library with the multi-pin technology |
|
1985 |
Limited peptide library using tea bag method |
|
1986 |
Iterative approach on solid phase peptide library screening using multi-pin Synthesis |
|
1986-90 |
Development of polynucleotide library method |
|
1988 |
Introduction of the split synthesis method on synthesizing a limited library of solution peptides |
|
1990 |
Light directed parallel peptide synthesis of a library of 1024 peptides on Chip |
|
1990 |
Successful application of the filamentous phase displayed peptide library method on a huge library of peptides |
|
1991 |
Introduction of the one bead- one compound concept and successful application of this concept to a huge bead- bound peptide library |
|
1991 |
Successful application of the iterative approach on a huge solution phase peptide library |
|
1992 |
Synthesis of a limited benzodiazepine- based small molecule library |
|
1992-93 |
Development of encoding methods for the one bead- one compound non- peptide library |
[Pandeya S.N. and Thakkar D. (2004)]16
EVOLUTION OF COMBINATORIAL CHEMISTRY
From a historical perspective, the research efforts made in classical combinatorial chemistry can be briefly outlined in three phases:
THE FIRST PHASE: In the early 1990s, the initial efforts in the combinatorial chemistry were driven by the improvements made in high-throughput screening (HTS) technologies. This led to a demand for access to a large set of compounds for biological screening. To keep up with this growing demand, chemists were under constant pressure to produce compounds in vast numbers for screening purposes. For practical reasons, the molecules in the first phase were simple peptides (or peptide-like) and lacked the structural complexity commonly found in modern organic synthesis literature.
THE SECOND PHASE: In the late 1990s, when chemists became aware that it is not just about numbers; but something was missing in compounds produced in a combinatorial fashion. Emphasis was thus shifted towards quality rather than quantity.
THE THIRD PHASE:
As the scientific community moved into the post-genomic chemical biology age, there was a growing demand in understanding the role of newly discovered proteins and their interactions with other bio-macromolecules (i.e. other proteins and DNA or RNA). For example, the early goals of the biomedical research community were centered on the identification of small-molecule ligands for biological targets such as G-protein-coupled receptors (GPCRs) and enzymes. However, the current challenges are moving in the direction of understanding bio-macromolecular (i.e. protein-protein, protein-DNA/RNA) interactions and how small molecules could be utilized as useful chemical probes in systematic dissection of these interactions. By no means will this be a trivial undertaking! The development of biological assays towards understanding bio-macro molecular interactions is equally challenging as the need for having access to useful small molecule chemical probes. [Kim S. (2005a)]9
PRINCIPLE OF COMBINATORIAL CHEMISTRY
The basic principle of combinatorial chemistry is to prepare a large number of different compounds at the same time Instead of synthesizing compounds in a conventional one at a time manner and then to identify the most promising compound for further development by high throughput screening (Process that allows rapid assessment of the activity of samples from a combinatorial library or other compound collection, often by running parallel assays in plates of 96 or more wells). The characteristic of combinatorial synthesis is that different compounds are generated simultaneously under identical reaction conditions (ie. using the same reaction conditions and the same reaction vessels.) in a systematic manner, so that ideally the products of all possible combinations of a given set of starting materials (termed building blocks) will be obtained at once. The collection of these finally synthesized compounds is referred to as a combinatorial library. The library is then screened for useful properties and the active compounds are identified.
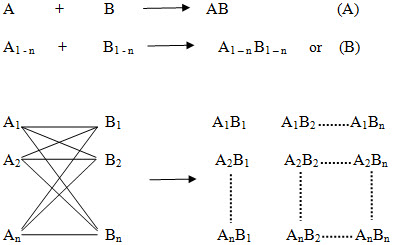
Figure -3. Principle of combinatorial chemistry (A) In general, in a conventional synthesis one starting material A reacts with one reagent B resulting in one product AB. (B) In a combinatorial synthesis different building blocks of type A (A1-An) are treated simultaneously with different building blocks of type B (B1-Bn) according to combinatorial principles, each starting material A reacts separately with all reagents B resulting in a combinatorial library A1-nB1-n.
The combinatorial libraries can be structurally related by a central core structure, termed scaffold (ie. all compounds of library have a common core structure), or by a common backbone. In both case, the accessible dissimilarities of compounds within the library depend on the building blocks which are used for the construction.
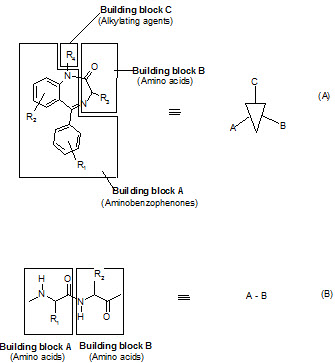
Figure -4. Combinatorial libraries (A) Scaffold based (B) Backbone based [Jung G. (1999)]8
TYPES OF COMBINATORIAL SYNTHESIS
Combinatorial chemistry is of two types:
(1)Solid phase combinatorial chemistry(The compound library have been synthesized on solid phase such as resin bead)
(2)Solution phase combinatorial chemistry (The compound library have been synthesized in solvent in the reaction flask)
(1) SOLID PHASE COMBINATORIAL CHEMISTRY
In solid phase combinatorial chemistry, the starting compound is attached to an insoluble resin bead, reagents are added to the solution in excess, and the resulting products can be isolated by simple filtration, which traps the beads while the excess reagent is washed away. [Leard L. & Hendry A. (2007)]1
The use of solid supports for chemical (non-peptides and peptide molecules) as well as biological synthesis (proteins, peptides, poly-nucleotides) relies/depends on three interconnected requirements:
a) A cross-linked, insoluble, polymeric material that is inert to the condition of synthesis.
b) Some means of linking the substrate to this solid phase that permits the cleavage of some or the entire product from the solid support during synthesis for analysis of the extent of reaction (s), and ultimately to give the final product of interest.
c) A chemical protection strategy (ie. Protecting group) to allow selective orthogonal protection and de-protection of reactive groups in the monomers. [Lather V. et al. (2005)]12
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Merrifield solid phase peptide synthesis:
Merrifield developed a series of chemical reactions that can be used to synthesize proteins. The direction of synthesis is opposite to that used in the cell. The intended carboxyl terminal amino acid is anchored to a solid support. Then, the next amino acid is coupled to the first one. In order to prevent further chain growth at this point, the amino acid, which is added, has its amino group blocked. After the coupling step, the block is removed from the primary amino group and the coupling reaction is repeated with the next amino acid. The process continues until the peptide or protein is completed. Then, the molecule is cleaved from the solid support and any groups protecting amino acid side chains are removed. Finally, the peptide or protein is purified to remove partial products and products containing errors.
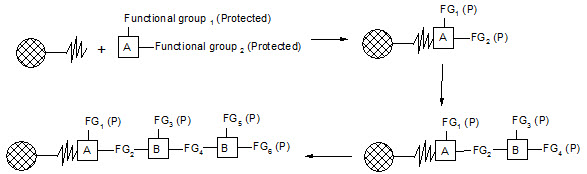
Figure-5. Solid-phase synthesis of peptides
combichemistry.com/solid_phase_synthesis.html3
Advantages of solid phase synthesis
(1) Since, the reaction is carried out on a solid support such as resin beads, a range of different starting materials are available that can be bound to separate resin beads, which are mixed together, such that all the starting material can be treated with another reagent in a single experiment. Therefore, it is possible to do multi-step synthesis and mix-and-split synthesis (a technique used to make very large libraries)
(2) Since, the products are bound to solid support, excess reagents or by-products can be easily removed by washing with appropriate solvent. Hence, large excesses of reagents can be used to drive reactions to completion.
(3) Intermediates in a reaction sequences are bound to the bead and need not be purified.
(4) Individual beads can be separated at the end of reaction to get individual products.
(5) The polymeric support can be regenerated and can be reutilized if appropriate cleavage conditions and suitable anchor/linker group are chosen. [Pandeya S.N. and Thakkar D. (2004)]16
(6) Because of easy separation of reagents and products, solid-phase chemistry can be automated more easily than solution chemistry. Separation of compounds bound to the solid support from those in solution is accomplished by simple filtration.
(7) Improved reaction yield. [Mishra A.K. et al. (2010)]15
Disadvantages of solid phase synthesis
(1) Optimal reaction conditions for solid phase synthesis can be difficult to determine, and developing these are far more time consuming than the actual reactions will be.
(2) There is limited the range of chemistry available for attachment to the resins in solid phase. ie. Some molecules don’t attach well to beads.
(3) It is difficult to monitor the progress of reaction when the substrate and product are attached to the solid phase.
(4) Assessment of the purity of the resin attached intermediates is also difficult.
(5) Purifying the final product after cleavage from the resin also proves to be a challenge. Removal of product from bead can be damaging to product if not careful. [Leard L. & Hendry A. (2007)]13
Solid support used in Solid phase synthesis
Most solid state combinatorial chemistry is conducted by using polymer beads ranging from 10 to 750 µm in diameter. The solid support must have the following characteristics for an efficient solid-phase synthesis:
1) Physical stability and of the right dimensions to allow for liquid handling and filtration;
2) Chemical inertness to all reagents involved in the synthesis;
3) An ability to swell while under reaction conditions to allow permeation of solvents and reagents to the reactive sites within the resin;
4) Derivatization with functional groups to allow for the covalent attachment of an appropriate linker or first monomeric unit. [Mishra A.K. et al. (2010)]15 The solid supports are usually composed of two parts: the core and the linker. The starting Compound of the synthesis is attached to the support via the linker.
Core--------Linker--------Start compound
The core ensures the insolubility of the support determines the swelling properties, while the linker provides the functional group for attachment of the start compound and determines the reaction conditions for the cleavage of the product. The linker itself and the covalent bond formed with the start compound must be stable under the reaction conditions of the synthesis. [Furka A. (2007)]5
Types of solid phase used:
* Polystyrene resins: In this, Polystyrene is cross linked with divinyl benzene (about 1% cross linking). Polystyrene resin is suitable for non-polar solvents.
* Tenta Gel resins: It consists of about 80% polyethylene glycol (PEG) grafted to cross-linked polystyrene1.It Combines the benefit of the soluble polyethylene glycol support with the insolubility & handling characteristics of the polystyrene bead. PEG containing resins are suitable for use in polar solvents.
* Poly acrylamide resins: Like super blue these resin swell better in polar solvent, since the contain amide bonds, more closely resemble biological materials.
* Glass and ceramic beads: These type of solid supports are used when high temperature and high pressure reaction are carried out.
[Mishra A.K. et al. (2010)]15
Linkers and anchors used in solid phase synthesis
The initial building block of the compound to be prepared by solid phase synthesis is covalently attached to the solid support via the linker. The linker is a bifunctional molecule. It has one functional group for irreversible attachment to the core resin and a second functional group for forming a reversible covalent bond with the initial building block of the product. The linker that is bound to the resin is called anchor.
Resin + Linker ----------> Resin----Anchor
The anchor can also be considered as a protecting group of one of the functional groups of the final product and, as such, it determines the reaction conditions by which the product can be cleaved from the support. A large variety of the commercially available resins contain the already built in anchor. A series of selected examples are found below.
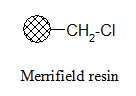
Merrifield resin:
The Merrifield resin can be used to attach carboxylic acids to the resin. The product can be cleaved from the resin in carboxylic acid form using HF.
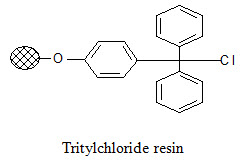
Tritylchloride resin:
The tritylchloride resin is much more reactive than the Merrifield resin. It can be used for attachment of a wide variety of compounds like carboxylic acids, alcohols, phenols, amines, thiols. The products can be cleaved under mild conditions using a solution of tri -fluoroacetic acid (TFA) in varying concentrations (2-50%).
Wang resin:
The resin is used to bind carboxylic acids. The ester linkage formed has a good stability during the solid phase reactions but its cleavage conditions are milder than that of the Merrifield resin.
Usually 95% TFA is applied. It is frequently used in peptide synthesis.
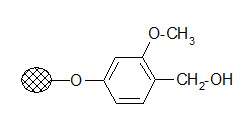
Wang resin
Hydroxymethyl resin:
The resin can be applied for attachment of activated carboxylic acids and the cleavage conditions resemble that of the Merrifield resin.
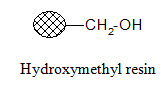
Aminomethyl resin:
Carboxylic acids in their activated form can be attached to the resin. Since the formed amide bond is resistant to cleavage, the resin is used when the synthesized products are not cleaved from the support; they are tested in bound form.
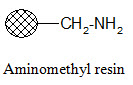
Rink amide resin:
The Rink resin is designed to bind carboxylic acids and cleave the product in carboxamide form under mild conditions. The amino group in the resin is usually present in protected form. For attachment of the substrate first the protecting group is removed then it is reacted with the activated carboxylic acid compound. Cleavage of the product in carboxamide form can be performed with dilute (~1%) TFA.
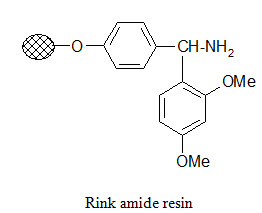
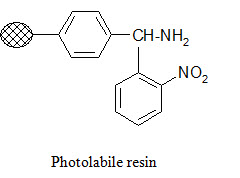
Photolabile anchors:
Photolabile anchors have been developed that allow cleavage of the product from the support by irradiation without using any chemical reagents. Such anchors, like the 2-nitro-benzhydrylamine resin below, usually contain nitro group that absorbs UV light.
Traceless anchors:
The initial building block of a multi-step solid phase synthesis needs to have one functional group (in addition to others) for its attachment to the solid support. It may happen that in t he end product t hi s group is unnecessary and needs to be removed. For this reason anchors have been developed that can be cleaved without leaving any functionality in the end product at the cleavage site. These traceless anchors usually contain silicon based linkers.
Protecting groups used in solid phase synthesis
If a chemist wants to carry out a reaction on only one functional group of a multifunctional group compound, the reactivity of the rest of the functional groups needs tobe suppressed. This can be achieved by application of protecting groups. A protecting group is reversibly attached to the functional group to convert it to a less reactive form. When the protection is no longer needed, the protecting group is cleaved and the original functionality is restored. A large number of protecting groups were developed for use in peptide synthesis since the amino acids are multifunctional compounds. It is an important requirement for a protecting group to be stable under the expected reaction conditions and to be cleavable - if possible-at mild reaction conditions. The stability/cleavage conditions of a protecting group are considered relative to those of the others. Two protecting groups are said to be orthogonal if either of them can be removed without affecting the stability of the other one. Some of the protecting groups most widely used in peptide synthesis are described below.
Protection of amino groups:
* The benzyl carbonyl (Z) group
Bergmann and Zervas suggested the benzyloxy carbonyl group for amino-protection in peptide synthesis in 1932 and this important protection type is still in use. The Z group can be introduced by the reaction of the amino group containing compound with benzylchloro- formate under Schott en-Bauman conditions.
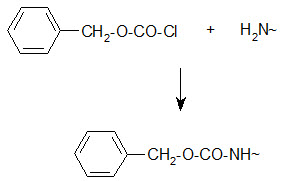
Protection of amino group by using benzyl carbonyl (z) group
The Z protection is stable under mildly basic conditions and nucleophilic reagents at ambient temperature. Cleavage can be brought about by HBr/AcOH, HBr/TFA or catalytic hydrogenolysis.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
* The t-butoxy carbonyl (Boc) group.
An alternative choice for amino group protection is the Boc group. Its advantage is that can be removed under milder conditions than the Z group.
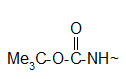
Protection of amino group by using t-butoxy carbonyl (Boc) group
The Boc group is completely stable to catalytic hydrogenolysis and as such is orthogonal to the Z group. Basic and nucleophylic reagents are no effect on the Boc group and its removal can be carried out by TFA at room temperature. The most convenient reagent that can be used in the protection reaction is the Boc anhydride (Boc2O).
The 9-fluorenyl methoxy carbonyl (Fmoc) group.
The Fmoc group differs from both Z and Boc groups since it is very stable to acidic reagents.
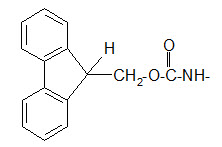
Protection of amino group by using 9-fluorenyl methoxy carbonyl (Fmoc) group
The Fmoc group can be removed under basic conditions. Usually 20% piperidine dissolved in DMF is used as reagent. One of the reagents for introducing the Fmoc group is the FmocCl.
Protection of carboxyl groups:
Carboxyl groups are most often protected by converting them to benzyl esters or t-butyl esters.
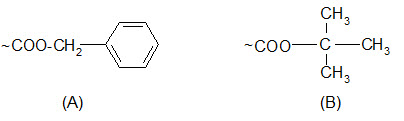
Protection of carboxyl groups by (A) benzyl esters and (B) t-butyl esters.
The benzyl esters are cleaved by saponification, HBr/AcOH, HF and catalytic hydrogenation but not by TFA. Their response to acids is similar to that of the Z groups but somewhat less sensitive.
The t-butyl esters, unlike benzyl esters, are stable to bases or nucleophilic attack. The properties of t-butyl esters are somewhat similar to those of the Boc groups although they are less sensitive to acidolysis. They can be cleaved by TFA.
Protection of other functional groups: The alcoholic and phenolic hydroxyl groups are protected by converting them to benzyl ether or t-butyl ether. The former protecting group can be cleaved by HF, HBr/AcOH or by catalytic hydrogenolysis and the latter one by TFA. Thiol groups can also be protected by benzyl ether formation or by tritylation.
The guanidino group (present in arginine) can be protected by nitration or by arylsulphonyl groups. The nitro group resists HBr/AcOH and can be cleaved by liquid HF. Among the arylsulphonyl groups the tosyl (Tos) group can be cleaved by liquid HF or sodium in liquid ammonia. Two other arylsulfonyl groups are more sensitive to acidic conditions.
The 2, 2, 5, 7, 8-pentamethylchroman- 6-sulphonyl (Pmc) group can be cleaved by TFA under conditions similar to the removal of the Boc group. The 4-methoxy-2, 3, 6 tri-methyl benzenesulphonyl (Mtr) group is also cleaved by TFA but is less sensitive and requires a few hours for cleavage.
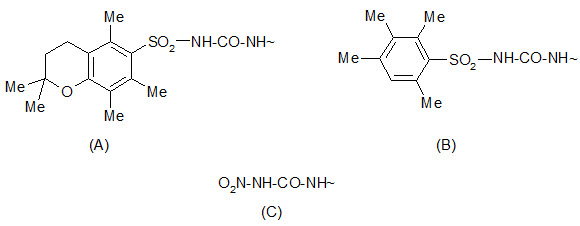
Protection by (A) Pmc group (B) Mtr group (C) Nitro group
The amide groups (in side chains of asparagine and glutamine) can be protected by tritylation. The trityl protecting group is stable to base, catalytic hydrogenolysis, very mild acid but is cleaved with TFA. It is used in conjunction with the Fmoc amino group protection strategy.
The NH group of the imidazole ring (in the side chain of histidine) is protected in conjunction with the Fmoc strategy by tritylation. The trityl protecting group can be removed by TFA at room temperature.
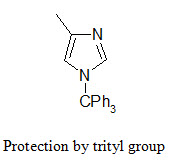
The indole ring (in tryptophane) can be protected by Boc group that can be removed by TFA.
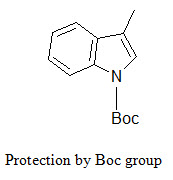
[Furka A. (2007)]5
(2) SOLUTION PHASE COMBINATORIAL CHEMISTRY
The solution phase synthesis involves conducting chemical reaction simultaneously, preferably in well-ordered sets (arrays) of reaction vessels in solution. [Pandeya S.N. and Thakkar D. (2004)]16 Most ordinary synthetic chemistry takes place in solution phase. The use of solution phase techniques has been explored as an alternative to solid-phase chemistry approaches for the preparation of arrays of compounds in the drug discovery process. Solution phase work is free from some of the constraints of solid-phase approaches but has disadvantages with respect to purification.
In solution phase synthesis we use soluble polymer as support for the product. PEG is a common vehicle which is used in solution phase synthesis it can be liquid or solid at room temperature and show varying degrees of solubility in aqueous and organic solvent. By converting one OH group of PEG to methyl ether (MeO-PEG-OH) it is possible to attached a carboxylic acid to the free OH and use in solution phase combinatorial synthesis. Another common support which is used in solution phase synthesis is liquid Teflon consisting mainly of long chain of (-CF2 -) groups attached to a silicon atom. When these phases are used as a soluble support for synthesis the resulting product can be easily separated from any organic solvent. [Mishra A.K. et al. (2010)]15
The main disadvantage of this method is when number of reagents are taken together in a solution, it can result in several side reactions and may lead to polymerization giving a tarry mass. Therefore, to avoid this, the new approach is developed in which all chemical structure combinations are prepared separately, in parallel on a giving building block using an automated robotic apparatus.
For example: Hundreds and thousands of vials are used to perform the reactions and laboratory robots are programmed to deliver specific reagents to each vial. [Pandeya S.N. and Thakkar D. (2004)]16
Table- 2. Characteristics of solid phase and solution phase combinatorial chemistry
|
SOLID PHASE |
SOLution PHASE |
|---|---|
|
Make a mixture of products |
Makes only one product |
|
Small amounts of products formed |
Large amounts of products formed |
|
Simple isolation of product by filtration |
Work-up and purification more difficult |
|
Requires two extra reaction steps: linkage & cleavage |
No extra steps for attachment & cleavage needed |
|
Limits to chemistry which can be performed |
Wide range of reactions can be utilized |
|
Automation possible |
Automation difficult |
|
Large excesses of reagent can be used to drive the reaction to completion |
Large excesses of reagent cannot be used as it causes subsequent separation problem. |
|
Longer reaction time than in solution phase |
Less reaction time |
|
Monitoring of reaction very difficult |
Monitoring of reaction easy |
|
Split and mix technique as well as parallel synthesis can be applied |
Split and mix strategy not possible Parallel synthesis can be applied |
[Leard L. & Hendry A. (2007)]13
SYNTHETIC METHODS USED IN COMBINTORIAL CHEMISTRY
There are two methods, which used for synthesis of compounds in combinatorial chemistry. They include:
(1) Split and mix synthesis or Split and pool synthesis or Portioning – Mixing (PM) synthesis (one bead-one compound library)
(2) Parallel synthesis (one vessel-one compound library)
(1) SPLIT AND MIX SYNTHESIS ‘OR’ PORTIONING–MIXING SYNTHESIS
This technique was pioneered by Furka and co-workers in 1988 for the synthesis of large peptide libraries. This approach is termed divide couple and recombine synthesis by other workers. [Jung G. (1999)]8
In this technique, the starting material is split in ‘n’ portions, reacted with ‘n’ building blocks, and recombined in one flask. For the second step, this procedure is repeated. This method is particularly employed for solid phase synthesis. [Pandeya S.N. and Thakkar D. (2004)]16
In following figure spheres represents resin beads, A, B & C represent the sets of building block and borders represents the reaction vessels. In the case, when three building blocks are used, in each coupling step after three stages (ie. divide, couple & recombine), a total number of 27 different compounds, one on each resin bead, are formed using 9 individual reactions (ignoring deprotection).
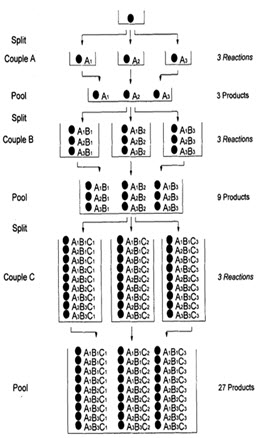
Figure- 6. Schematic diagram of split and mix synthesis [Jung G. (1999)]8
On the resulting products from split and pool synthesis, bioassays is performed and active mixture is discovered. Once an active mixture has been discovered, the next task is discovering which individual compound(s) in that mixture are active. The process of determining these active compounds is known as deconvolution.
Advantages:
a) Only few reaction vessels required
(b) Large libraries can be quickly generated (up to 105 compounds)
Disadvantages:
(a) Threefold amount of resin beads necessary
(b) The amount of synthesized product is very small.
(c) Complex mixtures are formed.
(d) Deconvolution or tagging is required.
(e) Synergistic effects may be observed during screening, leading to false positives. [Leard L. & Hendry A. (2007)]13
(2) PARALLEL SYNTHESIS
In this method, each starting material is reacted with each building block separately (ie. In separate vessel). After each reaction step the product is split into ‘n’ portions before it is reacted with n new building blocks. [Pandeya S.N. and Thakkar D. (2004)]16
In following figure spheres represents resin beads, A, B & C represent the sets of building block and borders represents the reaction vessels. In the case, when three building blocks are used, in each coupling step after three stages, a total number of 27 different compounds, one on each resin bead, are formed using 9 individual reactions (ignoring deprotection).
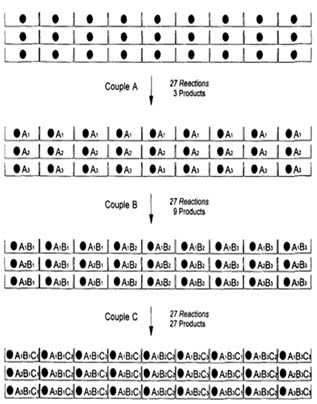
Figure-7. Schematic diagram of parallel synthesis [Jung G. (1999)]8
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Like the split and pool method, it results in the production of multiple compounds at the same time. However, unlike split and pool, parallel synthesis gives individual compounds, not a mixture. Thus deconvolution is not an issue in this method.
Advantages:
(1) It creates the compounds individually and in their own vessel. Thus the identity of the product is already known
(2) No deconvolution is required.
(3) Each compound is substantially pure in its location
(4) Defined location provides the structure of a certain compound
(5) Easier biological evaluation
Disadvantage:
(1) Applicable only for medium libraries (several thousand compounds)
(2) The amount of vessels required for this process is large, and the number of reactions performed is even greater.
[Leard L. & Hendry A. (2007)]13
[Pandeya S.N. and Thakkar D. (2004)]16
Methods for parallel synthesis
Methods that can be used for parallel synthetic includes:
(1) Multipin method
(2) Teabag method
(3) SPOTS membrane method
(1) Multipin method
This method was published by Geysen and his colleagues in 1984 for synthesizing a series of peptides epitopes by using Multipin apparatus. The multipin apparatus has a block of wells (96 wells format in 12×8 way) serving as reaction vessels and cover plate with mounted polyethylene rods or pins (4×40 mm) fitting into well. The end of polyethylene rods (pins) are coated with derivatized polyacrilic acid (marked by black).
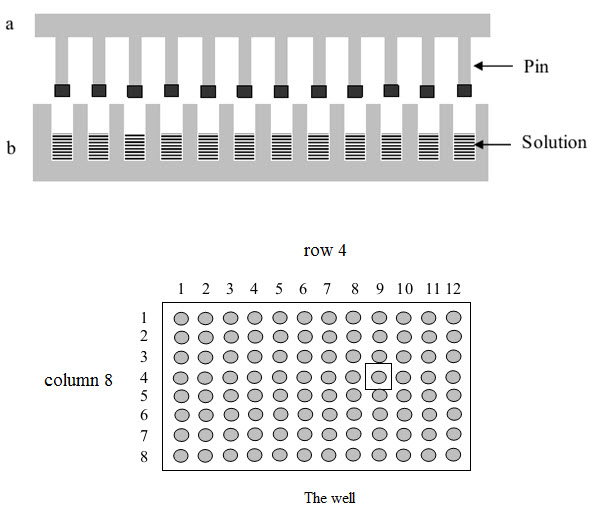
Figure-8. The Multipin apparatus
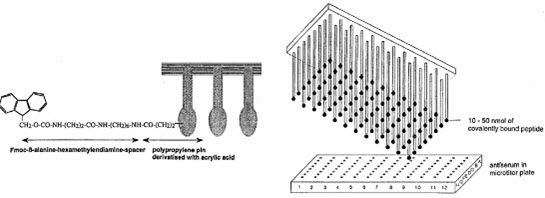
The protected amino acids used in building the peptides and the coupling reagents are dissolved and added to the wells. The coated ends of the pins are immersed into solution and kept there until the coupling reactions ended. The peptides formed on the pins immersed into solutions.
The sequence of peptides depended on the order of amino acids of added to the wells. [Furka A. (2007)]5
[Hima P. et al. (2009)]7
The peptides are screened by means of ELISA method after deprotection without leaving them from the pins to determine the binding capability of covalently bound peptide to antibodies. [Pandeya S.N. and Thakkar D. (2004)]16
The formed peptides are attached to the pins.The first amino acid was attached to the end of polyethylene rods (pins) grafted with derivatized polyacrylic acid (marked by gray). [Furka A. (2007)]5
[Hima P. et al. (2009)]7
[Kim S. (2005b)]10
The most characteristic feature is that the no. of product formed during the synthetic process never exceeds the no. of starting samples.
[Hima P. et al. (2009)]7
(2) SPOTS membrane method
This method introduced by Frank (1992) and his group also for preparing peptide arrays. The synthesis is carried out on cellulose paper membranes instead of polyethylene pins as the solid support for peptide synthesis. [Pandeya S.N. and Thakkar D. (2004)]16
Small droplets of solutions of protected amino acids dissolved in low volatility solvents & coupling reagents are pipetted on to predefined positions of the membrane. The spots thus formed can be considered as reactors when the conversion reactions of the solid phase synthesis take place.
Figure- 10. The SPOT synthesis
An array of as many as 2000 peptides can be made on a 8 x 12 cm paper sheet. The peptides can be screened on the paper after removing the protecting groups.
(3) Teabag method (Compromise between parallel & split - pool synthesis)
This method was developed in 1985 by Houghten for multiple peptide synthesis. The beads of the solid support are enclosed in permeable polypropylene (plastic) bags (5 x 22 mm mesh packets), then placed for coupling into a reaction vessel containing the solution of amino acid & the coupling reagent. All operations, including removal of protecting groups, couplings, washings & even the cleavages are performed on solid supports enclosed in bags. At the end of synthesis, each bag contains a single peptide. This procedure has significant advantages. [Hima P. et al. (2009)]7
(1) Greater Quantity of each compound is available at once (structural characterisation) (2) Labelling of the tea bags: Easier identification of each compound [Rose C. (2010)]19
All those bags which needed the attachment of the same amino acid (eg. Alanine) are grouped together, placed into the same reaction vessel,& the coupling can be done in a single operation. [Hima P. et al. (2009)]7
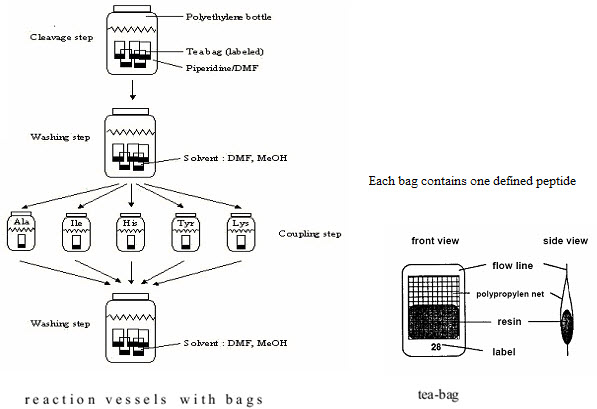
Figure-11. The Teabag synthesis
[Kim S. (2005b)]10
Miscellaneous methods for generating combinatorial libraries
The other approaches for generating combinatorial libraries includes :
(1) Light directed spatially addressable parallel chemical synthesis
(2) Biological method
(1) Light directed spatially addressable parallel chemical synthesis
A very remarkable combinatorial synthesis was developed by Fodor and his colleagues by combining the solid phase synthesis with the photolithographic procedure applied in the fabrication of the computer chips.
The method was published in 1991 under the title “The light-directed, spatially addressable parallel chemical synthesis”.
The light directed method makes it possible to prepare an array of peptides or other kinds of molecular on the surface of a glass slide. The surface of the glass is functionalized with amino alkyl groups protected by the photolabile 6–nitro Vera tryloxy carbonyl (NVOC) groups. The amino acid used in the synthesis are also protected by NVOC. group. [Furka A. (2007)]5
The basic principle of the method is that each set of building blocks contains a photolabile protecting group (NVOC) and only the building blocks which have been exposed to light can be coupled with another building block. Thus, pattern of masks and the sequence of protecting groups define the final structure of the compounds synthesized.

Figure- 12. Basic principle of light directed synthesis
[Rose C. (2010)]19
Example: Nine di-peptides are synthesized from amino acids A, G & K. Before each coupling step one or more area of the slide is irradiated through a mask to remove the protecting groups from those areas. Then the slide is submitted to coupling with the indicated protected amino acid. This can be done by immersing the slide into the solvent containing the protected amino acid and the coupling reagent. Although the full slide is submitted to coupling reaction, coupling occurs only in the irradiated area where the free amino acids are found. By completing a coupling cycle the full area of the slide becomes again protected. Before the next coupling cycle a new area have to be irradiated in order to produce free amino groups. The synthesis of 9 di-peptides is completed in 6 cycles irradiation and coupling (a to f).
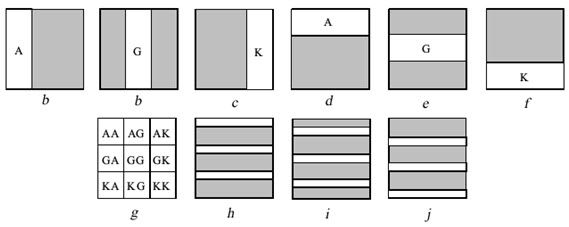
Figure- 13. The light – directed synthesis of nine dipeptides
[Furka A. (2007)]5
By irradiating through marks h, i, j & coupling with amino acids A, G, & K , 27 tripeptides will form. All these are individual compound which are formed is an efficient way resembling PM synthesis.
Advantages
The sequence are defined by their locations on the slide ie. Each member of the library is synthesized at a specific location.
(2) Biological method
In 1990 three different research groups introduced a new biological approach for producing peptide sequence libraries. This approach of creating peptide libraries is briefly exemplified by phage display libraries. First, an oligonucleotide library is synthesized chemically by a series of coupling with equimolar nucleotide mixtures. The formed oligonucleotides are then inserted into the DNA of phages. In the next stage the phages infect the host bacterium and their DNA replicate together with the inserted "foreign" DNA segment. A library of phages clone forms. Each clone carrier in its DNA a different "foreign" sequence segment which is expressed as a partial sequence of its coat protein. Every phage particle carries a couple of thousand identical coat protein molecules with the same peptides sequence fused to the Outer end. In this respect the phages resemble the bead in PM synthesis, with each containing an individual compound. The DNA of the phage can be considered as an encoding tag since the sequence of the peptide can be determined (after amplification) by sequencing the proper portion of the DNA. [Hima P. et al. (2009)]7
SCREENING OF COMBINATORIAL LIBRARIES
Screening is the process of determining whether compounds in a chemical library have a desired chemical or biological activity, without necessarily identifying the precise chemical nature of the compound(s) being screened. There are a range of options for testing the libraries in a biological assay. These include:
(i)Test mixture in solution: All the compounds are cleaved from the beads and tested in solution. If the resin beads were intimately mixed, it is not possible to test the products separately, but rather as a mixture. If activity in a pharmacological screen is observed it is not possible to say which compound or compounds are active. In order to identify the most active component, it is necessary to resynthesize the compounds individually and thereby find the most potent. This iterative process of resynthesis and screening is one of the most simple and successful methods for identifying active compounds from libraries.
(ii)Test individual compounds in solution: A second method is to separate the beads manually into individual wells and cleave the compounds from the solid-phase. These compounds can now be tested as individual entities.
(iii)Test compounds on the beads: A third method for screening is testing on the beads, using a colorimetric or fluorescent assay technique. If there are active compounds, the appropriate beads can be selected by color or fluorescence, ‘picked’ out by micromanipulation and the product structure, if a peptide, determined by sequencing on the bead. Non-peptide structures would need to be identified by one of the tagging methods. Screening on the bead may be an inappropriate method for drug discovery, as the bead and linker present conformational restrictions that may prevent binding to the receptor. Furthermore for pharmaceutical applications compounds will be invariably need to act, and thus ideally need to be tested in solution. [Leard L. & Hendry A. (2007)]13
ENCODING OF COMBINATORIAL LIBRARIES
By the process of screening the number of libraries that has “desirable properties” are sorted out. It is now very important to learn the identity of “winning” library member. The process of identification of active compound in a mixture of compounds is known as Encoding.
For identification of active compound following types of encoding methods are used:
(1) Positional encoding or deconvolution (iterative resynthesis and rescreening)
(2) Chemical encoding (Tagging)
(3) Electronic encoding
(1) Positional encoding or deconvolution (iterative resynthesis and rescreening)
In this method, the resynthesis and rescreening is carried out to know the identity of the active compound. In other terms, it is a process of optimizing an activity of interest by fractionating (normally by resynthesis, or by elaborating a partial library) a pool with some level of the desired activity to give a set of smaller pools. Repeating this strategy leads to single members with (ideally) a high level of activity and is termed iterative deconvolution. [Pandeya S.N. and Thakkar D. (2004)]16
In split-pool method, once active pools have been identified, and last step leading to these active pools is known. One now must work backwards to determine the order of reagent addition leading to the active compound(s). To do this, the reaction is begun a new, but when the last step is reached, the beads are not pooled and split, but separately reacted with the reagent that led to the active pool. These pools are now tested for activity. From the active pool found here, the second-last reagent added to give the active compound is now determined. This process is repeated again, until the order of reagents, and thus the active compound, has been identified.
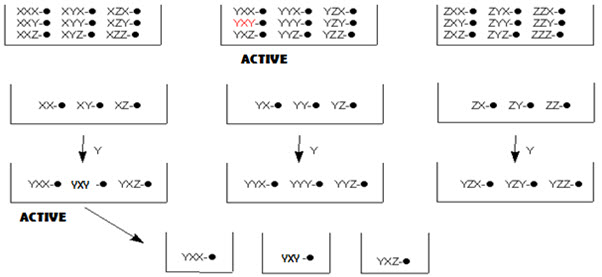
Figure- 14. Identification of active compound by iterative resynthesis and rescreening
This process is easiest when only a small number of compounds are expected to show any activity, as in the early stages of drug designs. Deconvolution is a time consuming method, taking longer than the original synthesis of the library. It also uses up reagents, thus it can also be an expensive process. Thus methods of tagging the resins, which allow for even simpler determination of the active compound, are generally employed now. [Leard L. & Hendry A. (2007)]13
(2) Chemical encoding (Tagging)
The most common approach to encoding solid phase libraries is to attach a chemical tag to the resin beads as the target molecule gets synthesized. Typically, at each step in the reaction, a tag is attached that is unique for the given step. For example, if we are creating a tripeptide and we have 10 possible amino acids at each position, we need to attach either a single tag that says “the tripeptide on this bead has amino acid Ala at position 1, Phe at position 2 and Gly at position 3” or we need to attach three different tags, one for each position. [Henry D.R. (2004)]6
After cleavage of the compound, the tags are cleaved (ie. decoding) from the solid support and analyzed by following analytical microtechniques:
* Polynucleotides (polymerase chain reaction-PCR), amino acids (Edman degradation & HPLC),
*Electrophoric tags (halocarbon molecules determined after silylation by gas chromatography),
* Amines (analyzed by HPLC) etc.
[Kom C. (2009)]11
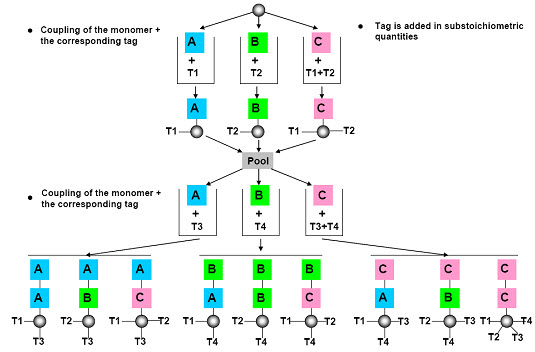
Figure- 15. Chemical encoding (tagging) [Kim S. (2005b)]10
(3) Electronic encoding
This technique uses a micro electronic device called a radio frequency (RF) memory tag. The tag measuring 13×3 mm is encased in heavy walled glass and contains the following:
- A silicon chip ,onto which laser etched a binary code,
- A rectifying circuit with which absorbed RF energy is converted to D.C. electrical energy,
- A transmitter/receiver circuit,
- An antenna, through which energy is received and RF signals are both received and sent.
[Pandeya S.N. and Thakkar D. (2004)]16
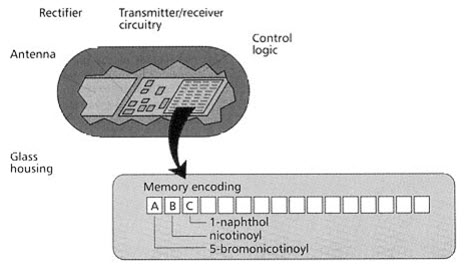
Figure-16. Radio frequency (RF) memory chip [Furka A. (2007)]5
THE APPLICATION OF COMBINATORIAL CHEMISTRY IN DRUG DISCOVERY
The combinatorial chemistry first shows its presence in synthesis of peptide libraries. The peptide plays varying role in body. By the use of combinatorial chemistry we can generate vast peptide, which may be active. Biologically active peptide hormones play an important role in regulating a multitude of human physiological response and many low molecular weight bioactive peptides can act as a hormone receptor against or antagonists. In addition, peptide structure commonly is found in molecules designed to inhibit enzymes that catalyze proteolysis, phosphorylation and other past translational protein modification that may play important role in pathologies of various disease states.
A few examples of the application of combinatorial chemistry in lead optimization and drug discovery are given below.
(a) Synthesis of peptoids
Some of the polypeptides of polypeptide libraries were found to be potent inhibitors for enzyme like kinases and proteases useful in treatment of AIDS and cancer, but these peptides have a poor bio-availability and unfavorable pharmacokinetic properties. So, the focus has been shifted on developing synthetic peptido mimetic like peptoides, one of the synthetic diversities has been developed by Simon et. al.
This group has created a basic set of monomers N-substituted glycine units, each bearing a nitrogen substitute similar to natural α-amino acid side chain. The formal polymerization of these monomers resulted in a class of polymeric diversity which was termed as ‘PEPTOIDS”. Peptoides may be synthesized either by “Full monomer” oligomers synthesis “Sub monomer” oligomers synthesis.
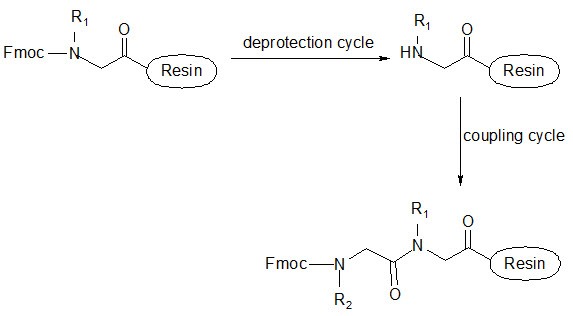
(a) Full monomer oligomers synthesis
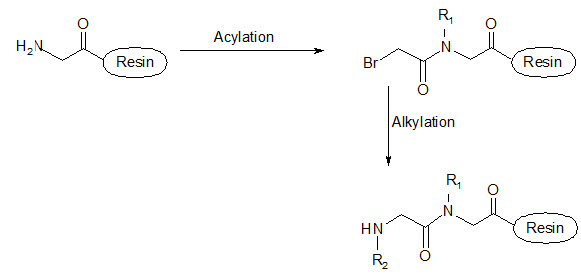
(b) Sub monomer oligomers synthesis
Figure- 17. Schematic representation of synthesis of peptoides
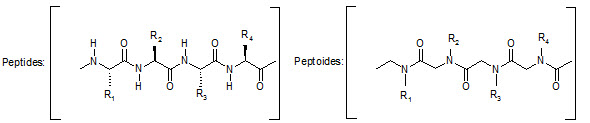
Comparison of peptide and peptoid backbone
Various biological activities have been established for specific peptoid synthesized, including inhibition of α-amylase and the hepatitis-A viruses 3C protease, binding to that fat RNA of HIV, antagonism at α1-adrenergic receptor.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
(b) Combinatorial lead optimization of a Neuropeptide-FF antagonist
Neuropeptide-FF (Phe-Leu-Phe-Gln-Pro-Gln-Arg-Phe-NH2) has been identified as a high affinity ligand for a G-protein coupled receptor HLWAR 77. It is an anti-opioid and has been implicated in pain modulation, morphine tolerance, and morphine abstinence. Centrally administered Neuropeptide-FF also has been known to precipitate Quasi- Morphine Abstinence Syndrome (QMAS) in opiate-naive animals. Therefore, antagonist of Neuropeptide-FF may allow easier management of withdrawal symptoms that adversely affect the treatment of opiate abuse.
Desaminotyrosyl-Phe-Leu-Phe-Gln-Pro-Gln-Arg-NH2, the first antagonist of Neuropeptide-FF was discovered but this analogue does not show any CNS bioavailability after systemic administration and, thus could not be considered as a potential lead compound. Derivatization with 5-(dimethylamino)-1-naphtha-lenesulfonyl (dansyl) at the secondary amino group of the N-terminal proline residue of the tripeptide Pro-Gln-Arg-NH2, obtained from the sequence of Neuropeptide-FF, has afforded an antagonist with significant lipid solubility to cross the BBB.
For a combinatorial optimizationto improve potency, libraries focused on the possible replacement of the proline and glutamine residues of this lead compound were obtained by a solid phase split and mix method using coded amino acid as building blocks. After screening for competitive binding against a radioiodinated Neuropeptide-FF analogue, 5-(dimethyl-amino)-1-naphthylene sulfonyl-Gly-Ser-Arg-NH2 (dansyl-GSR-NH2) has emerged as one of the compounds having high affinity to the Neuropeptide-FF receptor and over with a moderate increase in CNS penetration power compared to lead compound.
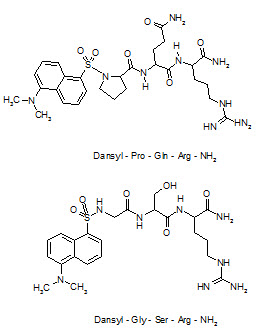
Structure of Neuropeptide-FF antagonist
(c) Generation of a benzodiazepine library
The seminar work of Ellman on solid phase synthesis of 1,4-benzodiazepine lays the ground work for creation of small molecules library and is considered as one of the most advancements in medicinal chemistry and represents the first example of the application of combinatorial organic synthesis to non polymeric organic compounds.
The benzodiazepines were synthesized on a solid support by the connection of three building blocks, and of different chemical families.
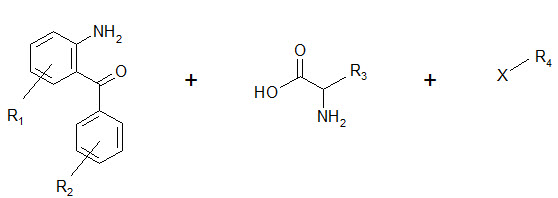
Components of a benzodiazepine library
Synthesis of benzodiazepine library:
1) Following the attachment of 2-aminobenzophenone hydroxy or carboxy derivative to the support using an acid cleavable linker [(N-hydroxymethyl) phenoxy-acetic acid], the N-protecting group is deblocked (piperdine/DMF).
2) The weak nucleophilic amine is acylated with an α-Fmoc-protected amino acid fluoride, using 4-methyl-2,6-di-tert-butylpyridine as an acid scavenger.
3) Fmoc deprotection followed by treatment with 5% acetic acid in DMF, causes the general cyclizationto the intermediate lactam.
4) Capitalizing on the ability of lithiated 5-(phenyl methyl)-2-oxazolidinone to selectively deprotonate the anilide NH2, alkylation was achieved with a variety of alkylating reagents. Aqueous acid cleaves the new benzodiazepine from the support in very high overall yields.
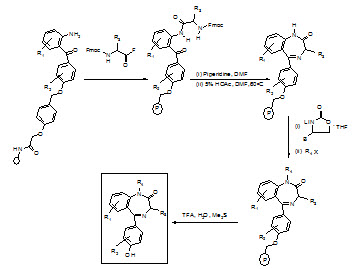
Figure- 18. Schematic representation of synthesis of benzodiazepin library
(d) Combinatorial lead optimization of Histamine H3 receptor antagonist
The H3 receptors are primarily located in the CNS in a presynaptic receptor that modulates the production and release of histamine. Blockade of this receptor leads to increased level of histamine and other neurotransmitters throughout the brain via effects on the pre and post synaptic H3 receptors. The wide distribution of H3 receptor in the mammalian CNS indicates a physiological role for this receptor. Therefore, its th erapeutic potential as a novel drug development target has been proposed for indications associated with neurological disorders such as Alzheimer’s disease, Parkinson’s disease and epilepsy, as well as metabolic disorders such as obesity.
A series of biaryl derivatives has been investigated to develop selective H3 blocker. A small library of 49 biphenyl-O-propylamine amides (a) were synthesized as singletons in solutions; the resulting products were purified using high throughput HPLC-MS techniques and assayed in a binding experiment using cloned human H3 and rat cortex H3 receptors. A number of potent inhibitors were found, one most potent being (b), was demonstrated excellent selective towards H3 receptors.
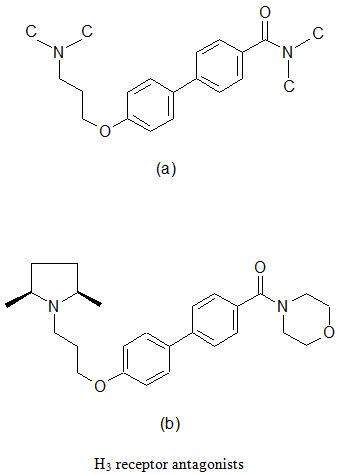
(e) Combinatorial lead optimization of dihydro-folate reductase inhibitor
The spread of antibiotic resistance has reached a larming proportions in some species, and one of the most worrying trends is the increasing incidence of methicillin resistant S.aureus in hospitals and multiresistant S. pneumoniae in the community. Therefore, there is an urgent need for effective antibacterial agents to treat infections caused by these organisms. The enzyme dihydrofolate reductase (DHFR) has been established in the clinic as a proven target for chemotherapy. The DHFR inhibitor Trimethoprim was introduced primary for the treatment of community-acquired infections and urinary track infections, with emphasis on gram-negative pathogens. The enzyme remains an under-exploited target in the antibacterial field and now optimization of inhibitors against gram-positive pathogens has been performed. Recent work has been conducted for improving the pharmacokinetic properties of DHFR inhibitors.
A library of 1392 compounds was synthesized in solution. The compounds were evaluated for inhibition of human DHFR and the bacterial enzymes from TMP-sensitive S. aureus and TMP-resistant S. pneumoniae . Several potent inhibitors were found, with one of the most potent compound (b) possessing IC50 value of 42 nM against S. aureus and 550 nm against S. pneumonia.
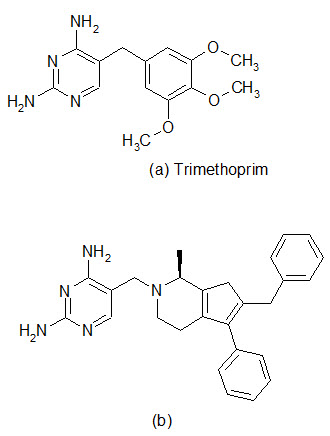
Dihydrofolate reductase (DHRF) inhibitors
[Pandeya S.N. and Thakkar D. (2004)]16
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
FUTURE OF COMBINATORIAL CHEMISTRY
The last ten years has seen an explosion in the exploration and adoption of combinatorial techniques. Indeed, it is difficult to identify any other topic in chemistry that has ever caught the imagination of chemists with such fervor.
For pharmaceutical chemists at least the reason for this change is not hard to fathom. 20 years ago the market for pharmaceuticals was growing at around 10% per annum but more recently the rate of the market growth as decline. At the same time, cost constraints on pharmaceutical research have forced the investigation of methods that offer higher productivity at lower expenses. The belief that combinatorial chemistry will allow the productive and cost-efficient generation of both compounds and drug molecules has fuelled enormous investment in this area.
Solid phase synthesis is highly suited to the synthesis of biopolymers such as DNA, RNA and peptides, as the chemistry required for chain extension is consistent for each step. It was therefore be worthwhile to put considerable effort into the optimization of the coupling conditions to give highly efficient syntheses. However, the history of drug discovery suggests that no single class of compound will provide all the drugs of the future, and thus for combinatorial chemistry to have maximum impact, a large range of bond-forming reaction need to be developed on solid phase.
However, much work remains to be done in this area and this is clearly an area of massive growth for the future.
Combinatorial chemistry represents a broad spectrum of techniques that are rapidly becoming a standard part of the medicinal chemist’s tool kit. But how will this technology develop in the future? Will it become a routine method of lead discovery used by all medicinal chemists or will it remain in the hands of specialists?
Whatever, the degree of integration into the medicinal chemist’s laboratory, one thing is certain. Combinatorial chemistry as a technique for the rapid synthesis of drug-like compounds will continue to make a major impact on the way drug molecules are discovered.
[combichemistry.com/conclusion.html]4
CONCLUSION
Combinatorial approaches have been introduced from the beginning in the drug discovery field, given their tremendous impact of the identification of new leads. Many active com-pounds have been selected to-date, following combinatorial methodologies, and a considerable number of those have progressed into clinical trials. However, combinatorial chemistry and related technologies for producing and screening large numbers of molecules also find useful applications in other industrial sectors not necessarily related to the pharmaceutical industry. Emerging fields of application of combinatorial technologies are diagnostics, the down-stream processing, catalysis and the new material sectors.
Many biotechnology/combinatorial-technology companies have been founded in the last few years, with the primary goal to design and produce highly diversified molecular libraries to be screened on selected targets, and the vast majority has definitely caught the attention of pharmaceutical companies. At the same time, rapidly growing sectors of catalyst design and new material design are going to influence chemical industries as well.
REFERENCES
1.Borman S. (1998). Combinatorial Chemistry. Chemical & Engineering News, American chemical society, Washington. April 6, pp. 47-67.
2.Combinatorial chemistry. [Internet]. Available at <combichemistry.com> [Accessed 27 December 2012]
3.Combinatorial synthesis on solid phase. [Internet]. Available at <combichemistry.com/solid_phase_synthesis.html> [Accessed 27 December 2012]
4.Future of combinatorial chemistry. [Internet]. Available at <combichemistry.com/conclusion.html> [Accessed 4 January 2013]
5.Furka A. (2007). Combinatorial chemistry: Principles and techniques. [Internet]. Available at <members.iif.hu/furka.arpad/BookPDF.pdf> [Accessed 10 January 2013]
6.Henry D.R. (2004). Combinatorial chemistry. In: Block J.H.& Beale J.M. (ed.). Wilson & Griswold’s, Text Book of Organic Medicinal & Pharmaceutical Chemistry. Lippincott Williams & Wilkins pp. - 52
7.Hima P. et al. (2009).Combinatorial chemistry. [Internet]. Available at <farmacists.blogspot.in/2009/05/combinatorial-chemistry.html> [Accessed 21 December 2012]
8.Jung G. (1999). Combinatorial Chemistry - Synthesis, Analysis, Screening. Wiley VCH, Weinheim, Germany, pp. 1 – 5
9.Kim S. (2005a). Introduction to combinatorial chemistry. Signal regulator synthesis lab. [Internet]. Available at <snupharm.ac.kr/shkim/erp/erpmenus/lesson_pds/upLoadFiles/L02%20Introduction%20to%20Combinatorial%20chemistry.pdf>[Accessed 25 December 2012]
10.Kim S. (2005b). Combinatorial chemistry. Signal regulator synthesis lab. [Internet]. Available at <http://www.snupharm.ac.kr/shkim/erp/erpmenus/lesson_pds/upLoadFiles/05.%20CombiChem.pdf> [Accessed 11 January 2013]
11.Kom C. (2009).Combinatorial chemistry : Active component identification, tagging. Wroctaw university of technology. [Internet]. Available at
<http://www.bioorganic.ch.pwr.wroc.pl/student/images/f/f8/ChKom8-9.pdf> [Accessed 11 January 2013]
12.Lather V. et al. (2005). An introduction to combinatorial chemistry: A rapid approach to lead development. [Internet]. Available at <http://www.pharmainfo.net/reviews/introduction-combinatorial-chemistry-rapid-approach-lead-development> [Accessed 26 December 2012]
13.Leard L. & Hendry A. (2007). Combinatorial chemistry in drug design. [Internet]. Available at <http://chem35132007.pbworks.com/w/page/15648417/Combinatorial%20Chemistry> [Accessed 4 January 2013]
14.Miertus S. et al. (2000).Concept of combinatorial chemistry and combinatorial technologies. Chem. Listy: The Official Journal of the Association of Czech Chemical Societies, 94, pp.1104 – 1110.
15.Mishra A.K. et al. (2010). Combinatorial chemistry and its application – a review. International journal of chemical and analytical science. 2010, 1(5), pp.100 – 105.
16.Pandeya S.N. and Thakkar D. (2004).Combinatorial chemistry: A novel method in drug discovery and its application. Indian journal of chemistry, Vol. 44B, February 2005, pp.335 – 348.
17.Patel R. (2008). Review on combinatorial chemistry & drug discovery. [Internet]. Available at <http://www.pharmainfo.net/pharma-student-magazine/review-combinatorial-chemistry-and-drug-discovery-0> [Accessed 27 December 2012]
18.Principle of combinatorial chemistry. [Internet]. Available at <combichemistry.com/principle.html> [Accessed 27 December 2012]
19.Rose C. (2010). Principles of combinatorial chemistry. [Internet]. Available at
<oc.chemie.uni-regensburg.de/OCP/ch/chv/oc22/script/002.pdf> [Accessed 23 December 2012]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






