
About Author
Ambika Shakya , Arabind kumar
Ambikashk9@gmail.com, arvindkumarshk@gmail.com
Kalka Institute For Research & Advanced Studies
NH-58, Baral, Partapur Bypass, Meerut-250103
Introduction
The disposition of a drug in the body involves absorption, distribution, metabolism, and excretion (ADME). ADME is an important part in the drug design process, which studies the upshot of a drug molecule after administration. It is a complex process involving transporters (Pgp) and metabolizing enzymes (CYP P450) with physiological consequences on pharmacological ( synergim and antagonism) and toxicological effects, and can play a major role in drug design for identifying better drug molecules in a more efficient way. Metabolism of drugs in the body is a complex biotransformation process where drugs are structurally modified to different molecules (metabolites) by various metabolizing enzymes. Studies on drug metabolism are key processes to optimize lead compounds for optimal Pharmacodynamic and pharmacokinetic properties, to identify new chemical molecules ( Zhang et al .,2018)
Drug metabolism occurs in many sites in the body main site of CYP meatabolism is the liver other sites is intestinal wall, lungs, kidneys, and plasma. The detoxification and excretion of xenobiotics (foreign drugs or chemicals) are occures in liver by enzymatically converting lipid-soluble compounds to more water-soluble compounds Xenobiotic is a term used to describe chemical substances that are foreign to animal life and thus includes such examples as plant constituents, drugs, pesticides, cosmetics, flavorings, fragrances, food additives, industrial chemicals and environmental pollutants. It has been estimated that humans are exposed to 1-3 million xenobiotics in their lifetimes Most of these chemicals that gain access to the body via the diet, air, drinking water, drug administration, and lifestyle choices, undergo a broad range of processes of detoxication that in general render them less toxic, more polar, and readily excretable (Patterson et al., 2010)
Pharmaceutical drugs, xenobiotics, as well as endogenous products are metabolized by a variety of cytochrome P450s (CYPs) that are widely distributed in the body. CYPs consist of numerous isozymes that have diverse metabolic roles and substrate specificities, each of which have a distinct effect on the rate of absorption and bioavailability of pharmaceutical drugs in the body. Products that alter P450-dependent drug metabolism, especially those that are not unajustable can therefore have a significant cascading impact on health (Scott et al., 2006).
Reference Id: PHARMATUTOR-ART-3003
The cytochrome P450 (CYP) enzymes are membrane-bound hemoproteins within cell, Induction or inhibitions of Cytochrome P450 e enzymes are major mechanism that underlies drug-drug interactions. CYP enzymes can be transcriptionally activated by variety of xenobiotics and endogenous substrates through receptor-dependent mechanisms. CYP enzyme inhibition by the many drugs is a major mechanism for metabolism- based drug-drug interactions. Many chemotherapeutic drugs can cause drug interactions due to their capacity to either inhibit or induce the CYP enzyme system (Palrasu et al., 2018).
Drugs that inhibit cytochrome P450 may increase plasma concentrations of other concurrently used drugs, resulting in drug interactions. Many other enzyme systems are inhibited by drugs, and enzyme inhibition may be the main mechanism of drug action. When two or more drugs are used concurrently, one drug may affect the action of the other (Calvey et al., 2005).
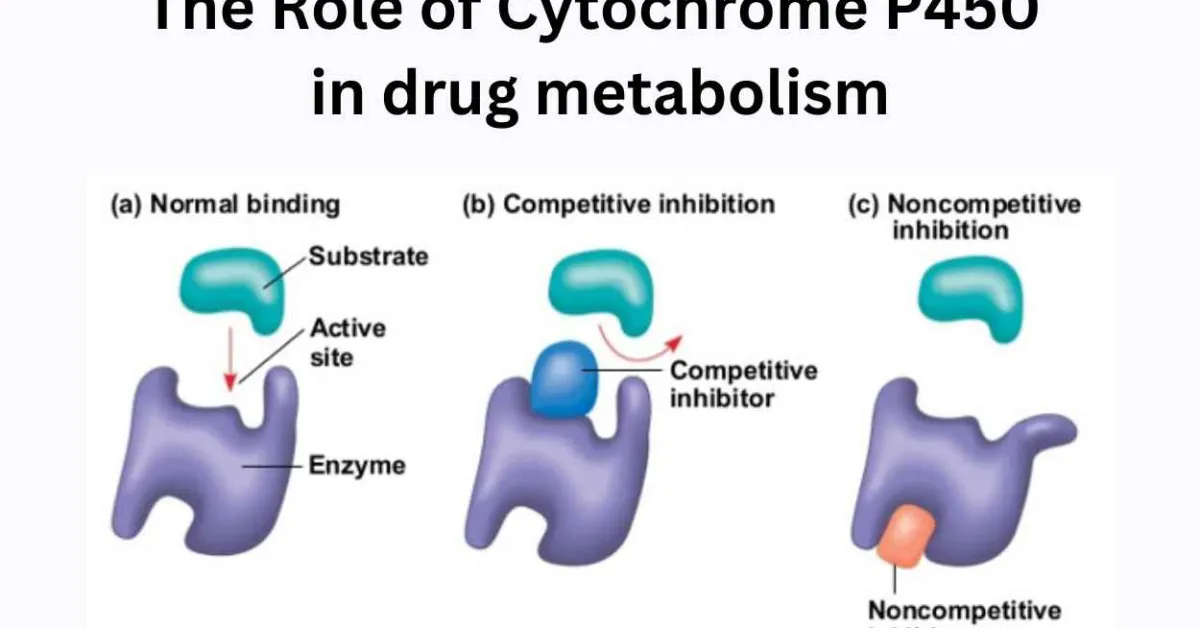
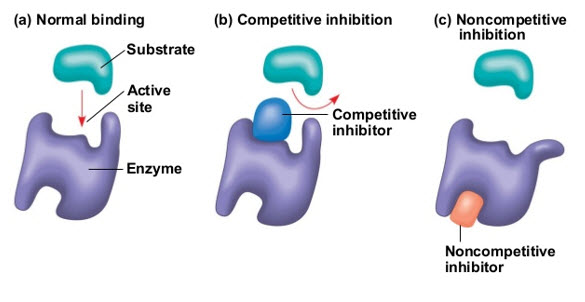
Enzyme inhibition and its type
Enzymes are proteins that help speed up metabolism, or the chemical reactions in our bodies. Enzymes are essential for digestion, liver function and much more our bodies naturally produce enzymes. But enzymes are also in manufactured products and food. The enzyme specificity is determined by the active site, a particular domain of the peptide molecule. There are two main theories looking for to explain the enzyme-substrate interaction. One – proposed by Fischer in the beginning of the 20th century – states that the substrate enzyme interaction would resemble a key-lock mechanism, in which the key (substrate) fits into the lock (active site), since two conditions are satisfied: the substrate and the active site have complementary structures and compatible polarity and size. The other – proposed by Koshland in the 1960 – states that the substrate nears the enzyme molecule induces on it some structural modifications that favor the enzyme-substrate fitting (Vitolo et al., 2019).
Two main type of inhibition 1.rversible 2.irreversible(or quasi-irreversible) other type is mixed type The enzyme inhibitors are molecules e. It has a hole (active site) in which a substance (substrate) can be transformed into another compound (product).which can bind to the enzyme and reduce the activity (Deodhar et al., 2020).
1. Reversible inhibition occurs as a result of competition at the active site of the enzyme and almost positively involves only the first step of the P450 catalytic cycle. Binding to the enzyme takes place usually with weak bonds, which are both formed and broken down easily. Consequently, reversible inhibitors act rapidly, but do not permanently destroy the enzyme competitive inhibition, competition between the substrate and inhibitor to bind to the same position on the active site of the enzyme takes place.
In the noncompetitive mode of inhibition, the active binding site of the substrate and inhibitor is different from each other. In practice, mixed-type inhibition displaying elements of both competitive and noncompetitive inhibition are frequently observed (Olavi et al., 2008) (Patadiya et al.,2021).
Cytochrome P450 inhibition and iduction with examples
Induction and inbition are basic mechanism of cytochrome P450 (CYP) enzymes ,by which drug–drug interactions ) such as mechanism-based inhibition. Each type of interaction involves a distinct clinical management strategy. Today, characteristics and regulatory factors of various CYP enzymes have been elucidated to a considerable extent .Detailed mechanisms of inhibition have been uncovered by studies on isolated or expressed enzymes and tissue fractions. Nuclear receptors as important xenobiotic-sensing transcription factors and as regulators of CYP induction have been elucidated (Hakkola et al ., 2020).
Frequent administration of certain drugs leads to increased synthesis (transcription or translation), or induction, of P450 enzymes. Enzyme induction increases metabolism of all drugs that are metabolized by that particular P450 isoenzyme. Therefore , when multiple drugs are given, drug interactions are likely at the level of P450 metabolism. This is the major mechanism for drug-drug interactions. For example, the antiepileptic drug phenytoin induces the CYP1A2 P450 isoenzyme. The antipsychotic drug haloperidol is metabolized by the same isoenzyme. If haloperidol is given concurrently with phenytoin, the metabolism of haloperidol will occur faster than normal as a result of enzyme induction, and the drug will be less effective. In this situation, practitioners may need to prescribe larger doses of haloperidol to achieve desired therapeutic effects. Haloperidol is commonly used in the therapy of patients with acute and chronic schizophrenia. The enzymes involved in the biotransformation of haloperidol include cytochrome P450 (CYP), carbonyl reductase and uridine diphosphoglucose glucuronosyltransferase. The greatest proportion of the intrinsic hepatic clearance of haloperidol is by glucuronidation, followed by the reduction of haloperidol to reduced haloperidol and by CYP-mediated oxidation (Mark et al., 2012) (Kudo et al .,1991).
In similarity to enzyme induction, some drugs block, or inhibit, the CYP enzymes that metabolize other drugs. The H2 (histamine) blocker cimetidine (used to treat acid reflux) is an example of a CYP2C9 P450 enzyme inhibitor in this case metabolism reduse and chances of increases of toxicity of other drug if given with cimetidine . Because diazepam (an anxiolytic drug ) is metabolized by the same CYP450 enzyme, when cimetidine is administered concurrently, diazepam will not be metabolized as rapidly as normal and may accumulate in the body the chances of toxicity of diazepam is more and this can lead to a longer t½ . the plasma level of substrates increases , can cross maximal effect of drug with coadministration of a P450 enzyme inhibitor and decreases with coadministration of a P450 enzyme inducer, with varying degrees of clinical significance. Natural and herbal products can also alter the activities of the microsomal P450 isoenzymes and alter drug metabolism To prevent adverse drug-drug interactions that occur as a concequences of altered metabolism (Mark et al ., 2012).
Enzyme induction causes major possessions on a limited number of extensively metabolized drugs (>75% ) metabolized) with a low therapeutic index.The enzyme induction is the de novo synthesis of new enzyme molecules resulting from an increased transcription of the respective gene after an appropriate stimulus. However, in drug metabolism research the term induction has been used as a generic term, describing the increase in the amount and/or activity of a drug metabolising enzyme as a result of an exposure to an inducing chemical irrespective of the underlying mechanism. Based on animal experiments, liver enzyme inducers have been classified mainly on the basis of the spectrum, i.e. the particular CYP family induced and the potency of the induction. For example, a typical enzyme inducer such as phenobarbitone will induce groups CYP1A, 2A, 2B and 3A, whereas alcohol is known to induce CYP2E1 The induction of an enzyme may result in also the inactivation or detoxification of an active of toxic substance or, in opposition, in the production of an active or toxic metabolite from an inactive precursor.( Sweenyet al., 2006) Drug-drug interactions have become an important issue in health care.
After coadministration administration of certain drugs leads to drug interaction of P450 enzyme. Enzyme induction increases metabolism of many drugs that are metabolized by that particular P450 isoenzyme and in case of inhibition decrese, metabolism of cuncurrent drug can lead to toxicity in body. Interactions can be explained by alterations in the metabolic enzymes that are present in the liver and other extrahepatic tissues.enzymes are being affected by previous administration of other drugs. After coadministration,some drugs act as potent enzyme inducers, whereas others are inhibitors (Andersonet al., 1998).
.Hormonal contraception, in plus the use of oral contraceptives (OCs), is widely used in many women with epilepsy – there is no strong evidence of seizures worsening with their use. Anti epileptic drugs (AEDs ) are the core for seizure control in women with epilepsy. on the other hand, there are many factors to consider in the choice of AED therapy and hormonal contraception, since some AEDs can reduce the efficacy of OCs having to pharmacokinetic interactions. Estrogens and progestogens are metabolized by cytochrome P450 3A4. AEDs, such as phenytoin, phenobarbital, carbamazepine, felbamate, topiramate, oxcarbazepine and primidone, induce cytochrome P450 3A4, leading to enhanced metabolism of either or both the estrogenic and progestogenic component of OCs, in that way reducing their efficacy in preventing pregnancy. OCs can also decrease the concentrations of AEDs such as lamotrigine and, thereby, increase the risk of seizures. Increased alertness of AED interactions may help optimize seizure therapy in women with epilepsy (Reddy et al. 2006).
Carbamezepine is an anticonvulsant medication used primarily in the treatment of epilepsy and neuropathic pain and also a unique example of an auto-inducer. Carbamazepine induces its own metabolism via CYP3A4, meaning that the dose of drug is given longer or increases because the drug is given, the more rapidly metabolized (Fuhr et al., 2020).
Rifampicin and isoniazid are first line drugs used in the treatment of tuberculosis, while rifampicin is highly effective in inducing hepatic, cyochrome P450 enzyme.
Rifampicin and isoniazid are main drugs used in the treatment of tuberculosis, while rifampicin is highly effective in inducing hepatic, drug metabolicP450 enzyme. When enzyme induction is achieved, the pharmacological effects of a specific drug may be reduced, since not only the metabolism of rifampicin itself, but also the metabolism of the other drug is accelerated The problem arises when doses are increased to reduce the effects of the combined drugs: increased serum concentrations of the combined drugs may possibly produce adverse effect because of the lost enzyme induction if rifampicin is discontinued. Rifampicin is also known to induce CYP3A4 and CYP2C9 (e.g. cyclosporin, diazepam andsteroids). As for dihydropyridine like nifedipine calcium channel blockers, it is somewhat possible that interactions with rifampicin may develop, since most of these drugs are metabolized by CYP3A4. food-drug interaction is an everyday occurrence, which can be bioavailability of a number of medications the mechanism of action, possible active ingredient in the juice and considers clinical implications. In 1989 it was reported that coadministration of grapefruit juice with the calcium channel antagonist felodipine resulted in a large increase in serum felodipine concentrations, as well as an enrichment of the pharmacodynamic effects of the drug Some drugs exhibit a significantly increased (up to three-fold) mean oral bioavailability when co-administered with grapefruit juice. reported that the inhibitory effect of grapefruit with ethanol and felodipine, it is 1,4-dihydropyridine calcium channel blocker Flavonoids (e.g. quercetin, naringenin, kaempferol) found up to 10% of the dry weight. It is thought that this naringin mainly inhibits the enzyme (CYP3A) that metabolizes calcium antagonists t½ increased. For example, interactions between benzodiazepines (e.g. midazolam, triazolam), antihistamines (e.g. terfenadine), immuno-suppressive drugs (e.g. cyclosporin) and grapefruit juice have been reported For example, (Hukkinen et al ,1995) studied 10 healthy young subjects who received a single 0.25 mg dose of triazolam with either 250 ml grapefruit juice or water. The plasma concentrations and effects of triazolam were measured up to 17 h. Grapefruit juice increased the AUC of triazolam in each subject and the Cmax in nine out of 10 subjects. The mean AUC of triazolam increased 1.5-fold (P < 0.001) and the peak concentration increased 1.3-fold (P < 0.05) following grapefruit juice. Grapefruit juice also postponed the peak time of triazolam from 1.6 to 2.5 h (P < 0.05). Grapefruit juice also increased the effects of triazolam, drowsiness being significantly (P < 0.05) enhanced.Other flavonoids (quercetin for example) may be major inhibitors of metabolism, the results of future studies are awaited with interest. It has already been established that, grape fruit juice is well known as potent inhibitors of cytochrome P450 3A4 in large amounts in oranges, grapefruit and their juices are known to alter the activity of P450 enzymes (P450 isoenzyme). The mechanism of inhibition of drug oxidation probably involves intestinal CYP3A4. The major grapefruit- specific flavonoid is naringin, which can account for up to 10% of the dry weight. It is believed that this naringin mainly inhibits the enzyme (CYP3A) that metabolizes calcium antagonists. For example, interactions between benzodiazepines (e.g. midazolam, triazolam), antihistamines (e.g. terfenadine), immuno-suppressive drugs (e.g. cyclosporin) and grapefruit juice have been reported (Bibi et al. 2008).
Grapefruit juice is consumed widely in today's health conscious world as a protector against cardiovascular diseases and cancers. Cardiovascular drugs constitute more than 50% of the close to 40 or more drugs so far known to interact with grapefruit .calcium channel blockers that have been discovered to interact with grapefruit include nifedipine, verapamil, diltiazem, nisoldipine, nimlodipine, nitrendipine, amlodipine and nifedipine. that the bioactive chemical compounds (furanocoumarins: bergamottin and 6′, 7′-dihydroxybergamottin) present in grapefruit inhibit intestinal CYP3A4 activity through a mechanism-based reaction, which causes degradation of the enzyme, hence reducing its levels by as much as 47% within four hours after grapefruit juice ingestion the interaction appears to compare with the oral bioavailability of these drugs, and that the interaction does not affect intravenously administered drugs led researchers to suspect as the main problem the intestinal cytochrome P450 (CYP3A4), which metabolises these drugs and many other (Kiani et al., 2007, Waghray et al., 2018).
The P – glycoprotein (Callaghan et al. 2014) is a transporte protein also known as multidrug resistance protein 1 (MDR1) found in the brush border of the intestinal wall which transports many of the cytochrome P – 450 3A4 substrates, has also been concerned to be inhibited by grapefruit juice. By inhibiting these enzyme systems, grapefruit juice alters the pharmacokinetics of a variety of medications, leading to elevation of their serum concentrations (Elimeliegy et al., 2020, .Linnet et al.2008).
Lots of of the drugs that are reported to interact with herbs are substrates of cytochrome (CYP) P450 enzymes; in particular CYP3A4 and CYP2C9, and these enzymes are subject to induction or inhibition by some of the regularly used herbal medicines. Accordingly, some of the herbal-drug interactions have been demonstrated to be mediated by CYP modulation, resulting in altered drug clearence and effect. On the other hand, expression of some of the CYP enzymes, including CYP3A4 is closely regulated by the nuclear factor pregnane X receptor (PXR) (Petr et al. 2026). A nuclear receptor protein that plays a critical role in the regulation of human cytochrome P450 x3A4 (CYP3A4) and transport protein (P glycoprotein) in Some of the drugs including cyclosporine, digoxine, simvastatin, imatinip, indinavir, saquinavir, among others, that interact with herbals are substrates for P-glycoprotein, and most of them are also substrates for CYP3A4, that is, they are dual substrates for both CYP3A4 and P-glycoprotein (P-gp). Like CYP3A4, P-gp can be induced or inhibited by drugs or herbals, and is also regulated by PXR. Thus, if a drug is a substrate for both CYPs and P-gp, there is a higher probability that it will interact with herbal products In fact, since herbal medicines are usually administered orally and may attain high concentrations in the gut lumen and the liver, they may significantly modulate the bioavailability of some orally administered drugs. There are many factors related to the patient, herb and the co-administered drug that may influence the clinical importance of drug interactions with herbs. For example, pharmacokinetic drug-herb interactions have more serious results with drugs with a narrow therapeutic range, such as warfarine or digoxin, since a small change in their plasma concentrations can lead to significant alterations in their therapeutic effect and/or toxicity. In most cases the extent of drug interactions with herbs varies markedly among individuals, depending on inter-individual differences in drug metabolizing enzymes and transport proteins, co-medication with other drugs, age, among many other factors Drugs that are identified to interact with herbals include anticoagulants and antithrombotic agents, cardiovascular drugs, immunosuppressants, sedatives and antidepressants, statins, anticancer drugs and anti-HIV agents. Most of the drugs that are known to interact with herbals are administered orally in long term treatments and many of them are substrates of cytochrome P450s (CYPs) and/or P-glycoprotein (P-gp). Herbal medicines that are reported to interact with drugs include garlic (Allium sativum), ginger (Zingiber officinale), ginkgo (Ginkgo biloba), ginseng (Panax ginseng), St. John’s wort, echinasea, valerian and aphedra (Dulger et al. 2012).
Conclusion
cytochromes P450 are isoenzyme containing heme act as a cofactor CYP450 so named as they bound to membranes with in cell that oxidise substances using iron involved drug metabolism and detoxification foreign chemicals and found high level in liver It has been estimated that 90%of drug oxidation can be attributed to six mainenzymes: CYP 1A2, 2C9, 2C19, 2D6, 2E1 and 3A4 The most significant CYP isoenzymes in terms ofquantity are CYP3A4 and CYP2D6. CYP3A4 is found not only in the liver but also in the gut wall Although this class has more than 50 enzymes, six of them metabolize 90 persent ofdrugs, , with the two most sinificant enzymes being CYP3A4 and CYP2D6. Cytochrome 450 enzymes can be inhibited and induced by drug,s , resulting in clinically significant drugs- drug interaction Cytochrome P450 enzymes being affected by previous administration of other drugs. After coadministration, some drugs act as potent enzyme inducers, while others are inhibitors. Drug interactions involving the P450 isoforms generally are of two types: enzyme induction or enzyme inhibition. When two drug are coadministerd cuncurrtntly, substrate for same enzyme second drug clear from the body without show pharmacological reaction and chances of therapeutic failure in case of induction .Enzyme inhibition reduces metabolism, whereas induction can increase
Drug interactions involving the P450 isoforms generally are of two types: enzyme induction or enzyme inhibition. Common substrates, inhibitors and inducers of P450 isozymes. Enzyme inhibition reduces metabolism, whereas induction can increase. The adverse effects can result in more than one way for example, from drug-drug interactions when one drug inhibits the metabolism of another drug increasing the drug to toxic levels.
References
1. Anderson GD ; (1998) ; “ A Mechanistics approach to antiepileptic drug interaction “ ; Ann Pharmacother ; 32 (5) ; 554-63
2. Bibi Zakia ; ( 2008) “ Retracted: Role of cytochrome P450 in drugInteractions “ ; Bibi Nutrition & Metabolism ; 5:27
3. Callaghan Richard ; Luk Frederick and Bebawy Mary ; (2014) ; “Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy?” ; Drug Metab Dispos.; 42(4): 623–631
4. Calvey Norrman (2005) ; “ Enzyme inducer and inhibitors addition substraction and synergism “ ; Anaesthesia & Intensive Care Medicine ; 6 ; 139 – 14
5. Deodhar Malavika; Al Rihani B Sweilem;Arwood J Meghan ; Darakjian Lucky;Dow pamela; turgeon Jacques and Michaud Veronique (2020);” Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice” ; Parmaceutics; 12(19); 846
6. Dulger Gul ( 2012) ; “ Herbal drugs and drug interactions “ ; Marmara Pharmaceutcal Journal ; 16: ; 9-22
7. Elimeliegy Mohamed ; Vourvahis Manoli ; Guo Cen ; Wang D. Diane ( 2020) ; “ Effect of P-glycoprotein (P-gp) Inducers on Exposure of P-gp Substrates: Review of Clinical Drug–Drug Interac1ion Studies ” ; Clin Pharmacokinet ; 59 (6) ; 699-714
8. Fuhr Maria Laura ; Marok Zahra Fatima ; Hanke Nina ; Selzer Dominik and Thorsten Lehr (2020) ; ‘A Mechanistic Approach to Antiepileptic Drug Interactions ‘ ; Pharnceutics
9. Hakkola Jukka;Hukkaren Jane; Turpeinen mila & Pelkonen Olavi (2020) ; “Inhibition and induction of CYP enzymes in humans: an update “ ; Archives of Toxicology ; 94 ; 3671–3722
10. Hukkinen SK, Varhe A, Olkkola KT, Neuvonen PJ: Plasma concentrations of triazolam are increased by concomitant ingestion of grapefruit juice. Clin Pharmacol Ther 1995, 58(2):127-131. 56
11. Kiani Jawad ; Imam Z Sardar ; (2007) “Medicinal importance of grapefruit juice and its interaction with various drugs “ ; Nutrition Journal ; 6 ; (2007)
12. Kudo S, Ishizaki T (1999 ),Pharmacokinetics of haloperidol: an update; Dec;37(6);435-
13. Linnet Kristian ; Ejsing Broeng Thomas ( 2008) ; “ A review on the impact of P-glycoprotein on the penetration of drugs into the brain. Focus on psychotropic drug “ ; Eur Neuro psychopharmacol ;. 18(3); 157-69
14. Mark Kester , Kent E. Vrana ( 2012) , in Elsevier's Integrated Review Pharmacology (Second Edition), 2012
15. Olavi Pelkonen; Miia Turpeinen; Jukka Hakkola; Paavo Honkakoski; Hukkanen Hukkanen; Hannu Raunio (2008); “Inhibition and induction of human cytochrome P450 enzymes: current statu” ; Arch Toxicol ; 82 ; 667–715
16. Palrasu Manikandan; siddavaram nagin (2018);”Naional Library Medicine Cytochrome P450 Structure, Function and Clinical Significance: A Review”; 19(1);38-54
17. Patadiya Nikunj ; Panchal Nikita; Vaghela Vipu ; (2021) ; “Mechanisms of CYP450 Inhibition: UnderstandingDrug-Drug Interactions Due to Mechanism-BasedInhibition in Clinical Practice “ ; A Review On Enzyme Inhibitors ; 12 (6)
18. Patterson D.Andrew; Frank J.gonzelew; Idle R. Jaffery (2010); “Xenobiotic Metabolism-A viewthought the metabolometer”; chemical research in toxicology; 17 ; 23(5) ; 851-860
19. Petr Pavek ( 2016 ) ; ( Prgnane X receptor ( PXR)- mediated gene repression and Cross – talk of PXR with Other Nuclear receptors Via Coactivator Interaction “ ; Front . Pharmacol
20. Reddy Samba Doodipala ; ( 2010) ; “Clinical pharmacokinetic interactions between antiepileptic drugs and hormonal contraceptives” ; Expert rev clin pharmacol ; 13(2):183-192
21. Scott I.M; Leduc R.I; Burt A.J; Marles R.J; Arnason ,J.T & Foster B.C ( 2006)”The Inhibition of Human Cytochrome P450 by Ethanol Extracts of North American Botanicals” ;Pharmaceutical Biology; 315-327
22. Sweeny B.P ; Bromilow J ( 2006) ; “ Liver Enzyme Induction and inhibition: implication for Anaesthesia “ ; Anaesthesia ; 61 ; 159-16l
23. Vitolo Michele;(2019);”brief review on enzyme activity”; World Journal of Pharmaceutical Research; 9(2) ;60-76.
24. Waghray Deepali ; Zhang Qinghai ; ( 2018) ; “ Inhibit or Evade Multidrug Resistance P-glycoprotein in Cancer Treatment “ ; J med chem ; 28; 61 (12)
25. Zhang Zhoupeng and Tang Wie (2018); “Drug metabolism in drug discovery development “; 8(5); 721–732








