
 About Authors:
About Authors:
Anoop Patel*1, Anoop Kumar1, Neha Sharma1, Monika Prajapati2
1Department of Pharmaceutical Technology, Meerut Institute of Engineering and Technology, Meerut, U.P., India
2Raj Kumar Goel institute of technology, 5th K.M. Stone Delhi Meerut Road, Ghaziabad, Uttar Pradesh- 201003
*anoop.p.2007@gmail.com
ABSTRACT:
Oral route still remains the favorite route of drug administration in many diseases because it is very suitable for drug delivery and non invasive. Till today it attracts to many researchers for investigation in the development of new dosage forms. The major problem in oral drug formulations is low and erratic bio-availability due to less water solubility and permeability of the drug across the biological membrane. This may arise high inter and intra subject variability due to lack of dose proportionality and therapeutic failure. It is estimated that 40% of new active constituents which are investigated recently show poor water solubility due to their lipophilic nature. The improvement of bio-availability of these drugs with such properties presents one of the greatest challenges in drug formulations. Several technologies are used for overcome these problems including micronization, solid dispersions, cyclodextrins complex formation and different lipid based drug delivery systems. Self-emulsifying drug delivery system is one the most important and advanced technology for enhancing the oral bio-availability as well reducing in dose. This system also gained attraction for enabling more consistent drug absorption, selective targeting of drugs in GIT, and protection of drugs from the inner environment of gut.
Reference Id: PHARMATUTOR-ART-1608
Introduction:
Still oral route is the most interested and favorable route for drug therapy. Many lipophilic approx 60% drugs manifest low oral bioavailability due to their poor aqueous solubility and low permeability through biological membranes. Having the low solubility and low permeability is major problem for the researchers. Amidon et al. classified these classes of compounds as low solubility/high permeability (class ?) and high solubility/low permeability (class III). Dissolution is the rate-controlling step in the absorption process, absorption and permeation across biological membranes both are important step for the proper bioavailabity [1]. Many Researches are still in progress to improve the oral bioavailability of lipophilic drugs in order to increase clinical effect of the API. Most popular approach is the incorporation of the active lipophilic compound into inert lipid vehicles [2], such as oils [3], surfactant dispersions [4, 5], self-emulsifying formulations [6, 7], emulsions [8, 9] and Liposomes [10]. Every formulation approach has its special advantages and limitations. Bioavailability of lipophilic drug is frequently obstructed due to their poor aqueous solubility and poor permeability leading to low absorption after in vivo administration. Plasma protein bound part of the administered dose is absorbed and reaches to the site of action and free part causes toxicity and undesirable actions due to unwanted biological distribution. Improved drug efficacy and less toxicity could be achieved through mixing of lipophilic drugs in lipids. The concept of drug delivery system has emerged to minimize the toxic side effects of drug, to broaden their application, to expand modes of their administration and to solve absorption problems. In the recent time remarkable growth is notice in drug development with the newly developed drugs. But the main problem is that the most compound are lipophilic with poor aqueous solubility and low permeability, which diminish their efficacy and bioavailability. Solubilization, encapsulation and lipid based formulations are the approaches to provide better absorption followed by lower dose, reduced frequency of administration, and improved therapeutic index.
Last two decades colloidal approaches [11-20] (liposomes, niosomes, microemulsion, organogels and nanocapsules) are used as vehicles in large scale for drug delivery. These self-built systems often lead to improve the therapeutic index of the lipophilic drugs through increased solubilization, permeation and modification of their pharmacokinetic profiles. For productive uses of this system in pharmacy, problems related to additives, stability over wide temperature range, low viscosity, small size biodegradability, and easy elimination from the body are some of the extremely important points. The size of the encapsulated particles should also very small because it may block the capillary so it needs to be reduce the size to control it; hence nano and micron-sized entities are preferred.
Development, characterization and biological studies of micro emulsion are very essential parameters to make them as potential vehicles for drug delivery [21-29] and it is vast area of research as they satisfy most of the required criteria [30-38]. Self-micro emulsifying drug delivery systems (SMEDDS) are isotropic mixtures of oils, surfactants and co-surfactant. Co-surfactants are used to produce fine oil-in-water emulsions when introduced into aqueous phase under gentle agitation [6, 7, 39, 40, 41]. In present time SMEDDS are formulating using medium chain tri-glyceride oils and nonionic surfactants. These are less toxic and suitable for lipid based formulations. Every oral administration emulsions (or micro-emulsions) form more fine emulsions in gastro-intestinal tract (GIT) due to gastric mobility it provides mild agitation to emulsion [42, 43]. Latent qualities of these systems are improved oral bio-availability, enabling reduction in dose, more consistent and better profiles of drug absorption, targeted delivery of drug in GIT and protection of drug(s) from the hostile environment in gut [44, 45]. The process of self-micro emulsification starts with the formation of liquid crystals (LC) and gel phases. Liquid crystals are responsible for the Release of drug from SMEDDS formed at the interface, since it is likely to affect the angle of curvature of the droplet and the resistance offered for partitioning of drug into aqueous media [46]. Effect of LC will be particularly noticeable for semisolid or solid SMEDDS because LC phases are formed in-situ, and the drug diffuses through LC phases into aqueous media. In the present topic, focus will be on lipid based drug delivery systems (e.g. Self-micro emulsifying Drug Delivery systems). Emulsion particles can be of either micro- or nano- size, depending on the composition of the system. These formulations circumvent the dissolution step in the gastro-intestinal tract, but are still dependent on digestion.
SMEDDS form transparent micro emulsions with a droplet size of less than 50 nm also the concentration of oil in SMEDDS is less than 20%. SMEDDS are physically stable formulations that are easy to manufacture [47].
Microemulsion [48]:
This approach is used for developing the formulation of hydrophobic agents for oral delivery. Like the other emulsion formulations microemulsion is also liquid dispersion of oil in water, stabilized by surfactants and co-surfactants. The microemulsion particles are smaller than the normal emulsion so the microemulsions essentially clear. Micro emulsions are isotropic mixture however are thermodynamically stable and are not subject to the particle agglomeration problems of conventional emulsions. It is generally noticed that micro emulsions are micelle-like particles, having an micellar structure that containing a distinct oil phase in the micelle core. These micelle like particles are often referred as swollen micelles, a term which emphasizes their close relationship to true micellar particles. In spite of their close relationship to micelles, microemulsion functions quite differently in drug delivery systems. Hydrophobic agents are generally lipophilic in nature and have a greater solubility in triglycerides than in surfactants. As a result, the hydrophobic therapeutic agent in microemulsion-based delivery system is preferentially solvated in triglyceride phase, which in turn encapsulated in the swollen micelle. Loading dose is depend on partitioning in the triglyceride phase than in comparable micelle-based systems but in these delivery systems the lipolysis dependency is the major disadvantage. Larger size of microemulsion particles results in a slower rate of particle diffusion and thus slower rate of therapeutic agent absorption. Thus there is a need for pharmaceutical compositions which have the property to overcome these limitations of conventional micelle formulations, but without having these disadvantages of triglyceride.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Oils [49]
Long chain triglyceride and medium chain triglyceride oils with different degree of saturation have been used in the design of SMEDDS. Unmodified edible oils provide the most natural basis for lipid vehicles, but their poor ability to dissolve large amounts of hydrophobic drugs and their relative difficulty in efficient self?micro emulsification markedly reduces their use in SMEDDS. Recently medium chain triglycerides are replaced by novel semi synthetic medium chain triglycerides containing compound such as GELUCIRE, Other suitable oil phases are digestible or non?digestible oils and fats such as olive oil, corn oil, soya bean oil, palm oil and animal fats etc.
Surfactants [50]
Various non?ionic surfactants such as the polysorbates and polyoxyls, which cover the HLB range from 2 to 18, may be used in combination with lipid excipients to promote self?emulsification or micro?emulsification.Due to their relatively low toxicity, the acceptable quantities for use of these surfactants are limited primarily by their tendency, at high concentration, to brittleness of hard and soft gelatin capsules due to their dehydrating effects on capsule gelatin. Surfactant have a high HLB & hydrophilic which assist the immediate formation of O/W droplet & rapid spreading of the formation in aqueous media. Surfactants are amphilic in nature & they can dissolve or soluble relatively high amount of hydrophobic drug compound. This can prevent precipitations of the drug within the GI lumen and for prolong existence of drug molecules. Due to their relatively low toxicity, the acceptability quality for use of these surfactant are limited primarily by their tendency, at high concentration, to cause brittleness of hard & soft gelatin capsule due to their dehydrating effect on capsule gelatin.
Cosolvents [51]
Co-solvents like ethanol, propylene glycol, polyethylene glycol, polyoxyethylene, propylene carbonate, tetrahydrofurfuryl alcohol polyethylene glycol ether (Glycofurol), etc., may help to dissolve large amounts of hydrophilic surfactants or the hydrophobic drug in the lipid base. These solvents sometimes play the role as co-surfactant in the micro-emulsion systems.
Need of SMEDDS
Oral delivery of Class II and Class III compounds is done by to fill the formulation into soft gelatin or hard gelatin capsules in addition of pre-dissolve compound in a suitable solvent. The main benefit of this approach is that pre-dissolve compound overcomes the initial rate limiting step of particulate dissolution in the aqueous environment within the GI tract. However, a potential problem is that the drug may precipitate in the solution when the formulation disperses in the GI tract, particularly if a hydrophilic solvent is used (e.g. polyethylene glycol). If the drug can be dissolved in a lipid vehicle there is less potential for precipitation on dilution in the GI tract, as partitioning kinetics will favor the drug remaining in the lipid droplets [52]. Another strategy for poorly soluble drugs is to formulate in a solid solution using a water-soluble polymer to aid solubility of the drug compound. For example, poly vinyl pyrrolidone (PVP) and polyethylene glycol (PEG 6000) have been used for preparing solid solutions with poorly soluble drugs. One potential problem with this type of formulation is that the drug may favor a more thermodynamically stable state, which can result in the compound crystallizing in the polymer matrix. Therefore the physical stability of such formulations needs to be assessed using techniques such as differential scanning calorimetry or X-ray crystallography. In this type of case SEDD system is a good option. Potential advantages of these systems include;
1. Enhanced oral bioavailability enabling reduction in dose,
2. More consistent temporal profiles of drug absorption,
3. Selective targeting of drug(s) toward specific absorption window in GIT,
4. Protection of drug(s) from the hostile environment in gut.
5. Control of delivery profiles
6. Reduced variability including food effects
7. Protective of sensitive drug substances
8. High drug payloads
9. Liquid or solid dosage forms
Mechanism of Self-Emulsification
The process of self-emulsification is not yet well understood. However, according to Reiss [53], self-emulsification occurs when the entropy change that favors dispersion is greater than the energy required to increase the surface area of the dispersion. In addition, the free energy of a conventional emulsion formation is a direct function of the energy required to create a new surface between the two phases and can be described by equation [53].
ΔG=∑ Niri2σ
Where, G is the free energy associated with the process (ignoring the free energy of mixing), N is the number of droplets of radius r and σ represents the interfacial energy. With time, the two phases of the emulsion will tend to separate with the reduction in the interfacial area and the free energy of the systems.
Therefore, the emulsions resulting from aqueous dilution are stabilized by conventional emulsifying agents, which form a monolayer around the emulsion droplets, and hence, reduce the interfacial energy, as well as providing a barrier to coalescence. In the case of self-emulsifying systems, the free energy required to form the emulsion is either very low and positive, or negative (then, the emulsification process occurs spontaneously). Emulsification requiring very little input energy involves destabilization through contraction of local interfacial regions. For the emulsification process it is necessary to have no resistance to surface shearing of the interfacial structure [54]. In earlier work, it was suggested that the ease of emulsification could be associated with the ease by which water penetrates into the various LC or gel phases formed on the surface of the droplet [6, 55, 56]. According to Wakerly et. al. [6] the addition of a binary mixture (oil/non-ionic surfactant) to water results in interface formation between the oil and aqueous-continuous phases, followed by the solubilization of water within the oil phase owing to aqueous penetration through the interface. This will occur until the solubilization limit is reached close to the interface. Further aqueous penetration will result in the formation of the dispersed LC phase. As the aqueous penetration proceeds, eventually all material close to the interface will be LC, the actual amount depending on the surfactant concentration in the binary mixture. Once formed, rapid penetration of water into the aqueous cores, aided by the gentle agitation of the self-emulsification process, causes interface disruption and droplet formation. The high stability of these self-emulsified systems to coalescence is considered to be due to the LC interface surrounding the oil droplets. The involvement of the LC phase in the emulsion formation process was extensively studied by Pouton et al. [6,56,57,58]. Later, Craig et al. used the combination of particle size analysis and low frequency dielectric spectroscopy (LFDS) to examine the self-emulsifying properties of a series of Imwitor 742 (a mixture of mono- and diglycerides of capric and caprylic acids)/Tween 80 systems [7,41,59]. The dielectric studies provided evidence that the formation of the emulsions may be associated with LC formation, although the relationship was clearly complex [59]. The above technique also pointed out that the presence of the drug may alter the emulsion characteristics, possibly by interacting with the LC phase[41]. However, the correlation between the spontaneous emulsification and LC formation is still not definitely established [41, 60].
General formulation approach
Preliminary studies are performed for selection of oil, which is an important and critical requisite for formulation of SEDDS. SEDDS consisted of oil, a surfactant and a co-surfactant. Solubility of drug is determined in various oils and surfactants. Prepare a series of SEDDS system containing drug in various oil and surfactant. Then, in vitro self-emulsification properties and droplet size analysis of these formulations upon their addition to water under mild agitation conditions is studied. Pseudo-ternary phase diagram is constructed, identifying the efficient self-emulsification region. From these studies, an optimized formulation is selected and its bio-availability is compared with a reference formulation [45].
The efficiency of oral absorption of the drug compound from the SEDDS depends on many formulation-related parameters, such as surfactant concentration, oil/surfactant ratio, polarity of the emulsion, droplet size and charge, all of which in essence determine the self-emulsification ability. Thus, only very specific pharmaceutical excipient combinations will lead to efficient self-emulsifying systems.
SMEDDS are differentiated from SEDDS by transparent or translucent solution with smaller emulsion droplets produced on dilution. SMEDDS generally contain relatively high concentrations of surfactant (typically 40-60% w/w), and regularly contain hydrophilic co-solvents (e.g. propylene glycol, polyethylene glycols) and low concentration of oil (20%). They are often described as micro-emulsion formed on dilution in aqueous media [61] When developing lipid based formulations the following parameters are believed to be important;
• The solubility of drug in the formulation as such and upon dispersion (for SEDDS),
• The rate of digestion (for formulations susceptible to digestion) and possibly
• The solubilization capacity of the digested formulation
Oils
Both long- and medium-chain triglyceride (MCT) oils with different degrees of saturation have been used for the design of self-dispersing formulations. Unmodified edible oils provide the most `natural' basis for lipid vehicles, but their poor ability to dissolve large amounts of hydrophobic drugs and their relative difficulty in efficient self-emulsification markedly reduce their use in SEDDS. In contrast, modified or hydrolyzed vegetable oils have contributed widely to the success of the above systems [40, 62, 63]. So they exhibit formulative and physiological advantages. These excipients form good emulsification systems, with a large number of non-ionic surfactants approved for oral administration, while their degradation products resemble the end products of intestinal digestion. MCTs were preferred in the earlier self-emulsifying formulations [39, 64]. Because of higher Fluidity, better solubility properties and self-emulsification ability, but evidently, they are considered less attractive compared to the novel semi-synthetic medium chain derivatives [40] which can be defined rather as amphiphilic compounds exhibiting surfactant properties. In such cases, the more lipophilic surfactant may play the role of the hydrophilic oil in the formulation [40,43].Solvent capacity for less hydrophobic drugs can be improved by blending triglycerides with mono- and di-glycerides[45].
Surfactants
Non-ionic surfactants with a relatively high hydrophilic± lipophilic balance (HLB) were advocated for the design of self-dispersing systems, where the various liquid or solid ethoxylated polyglycolyzed glycerides and polyoxyethylene 20 oleate (Tween 80) are the most frequently used excipients. Emulsifiers derived from natural sources are expected to be safer than synthetic ones and are recommended for SDLF (self dispersed lipid formulation) use [40,63,65,66], in spite of their limited ability to self-emulsify. Non-ionic surfactants are known to be less toxic compared to ionic surface-active agents, but they may cause moderate reversible changes in intestinal wall permeability [6, 67]. Amemiya et al. proposed a new vehicle based on a fine emulsion using minimal surfactant content (3%) to avoid the potential toxicological problems associated with high surfactant concentration [68]. The usual surfactant concentration in self-emulsifying formulations required to form and maintain an emulsion state in the GI tract ranged from 30 to 60% w/w of the formulation. A large quantity of surfactant may irritate the GI tract. Thus, the safety aspect of the surfactant vehicle should be carefully considered in each case. The high HLB and subsequent hydrophilicity of surfactants is necessary for the immediate formation of o/w droplets and/or rapid spreading of the formulation in the aqueous environment, providing a good dispersing/self emulsifying performance. The surface-active agents are amphiphilic by nature, and they are therefore usually able to dissolve and even solubilize relatively high quantities of the hydrophobic drug. The latter is of prime importance for preventing precipitation within the GI lumen and for the prolonged existence of the drug molecules in soluble form, which is vital for effective absorption [64]. The lipid mixtures with higher surfactant and co-surfactant/oil ratios lead to the formation of self-micro emulsifying formulations (SMEDDS) [40,69,70,71]. Formulations consisting only of the surfactant mixture may form emulsions or microemulsions (when surfactants exhibit different low and high HLB) [43],micelle solution or, in some particular cases, niosomes, which are non-ionic, surfactant-based bilayer vehicles [72].
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Co-solvents
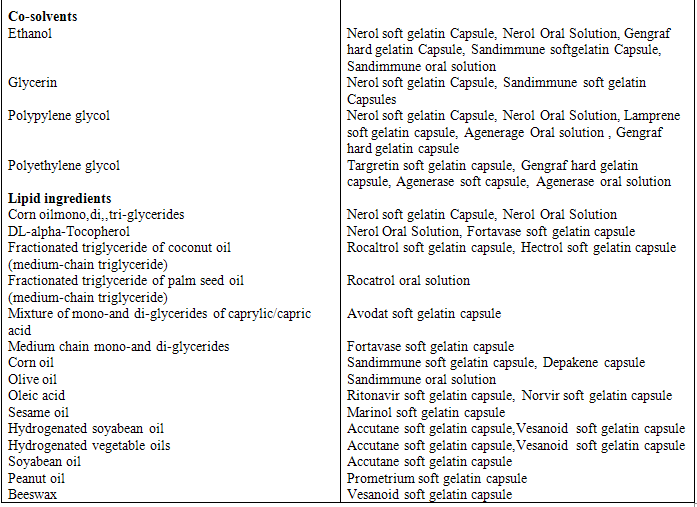
Relatively high surfactant concentrations (usually more than 30% w/w) are need in order to produce an effective self-emulsifying system. Organic solvents, suitable for oral administration (ethanol, propylene glycol (PG), polyethylene glycol (PEG), etc. may help to dissolve large amounts of either the hydrophilic surfactant or the drug in the lipid base. These solvents sometimes play the role of the co-surfactant in the micro emulsion systems, although alcohol-free self-emulsifying microemulsions have also been described in the literature [40]. Indeed, such systems may exhibit some advantages over the previous formulations when incorporated in capsule dosage forms, since alcohol and other volatile co-solvents comprised in the conventional self-emulsifying formulations are known to migrate into the shells of soft gelatin, or hard, sealed gelatin capsules, resulting in the precipitation of the lipophilic drug. On the other hand, the lipophilic drug dissolution ability of the alcohol free formulation may be limited. Drug release from the formulation increased with increasing amount of co-surfactant. Various examples of surfactant, co-solvents and oil are given in table 1.
Table 1 Example of surfactants, co-surfactant, and co-solvent used in commercial formulations

EVALUATION:
Thermodynamic stability studies
The physical stability of a lipid based formulation is also crucial to its performance, which can be adversely affected by precipitation of the drug in the excipient matrix. In addition, poor formulation physical stability can lead to phase separation of the excipient, affecting not only formulation performance, but visual appearance as well. In addition, incompatibilities between the formulation and the gelatin capsules shell can lead to brittleness or deformation, delayed disintegration, or incomplete release of drug.
1. Heating cooling cycle: Six cycles between refrigerator temperature (40C) and 450C with storage at each temperature of not less than 48 h is studied. Those formulations, which are stable at these temperatures, are subjected to centrifugation test.
2. Centrifugation: Passed formulations are centrifuged thaw cycles between 21 0C and +25 0C with storage at each temperature for not less than 48 h is done at 3500 rpm for 30 min. Those formulations that does not show any phase separation are taken for the freeze thaw stress test.
3. Freeze thaw cycle: Three freeze for the formulations. Those formulations passed this test showed good stability with no phase separation, creaming, or cracking [73].
Dispersibility test
The efficiency of self-emulsification of oral nano or micro emulsion is assessed using a standard USP XXII dissolution apparatus 2. One milliliter of each formulation was added to 500 mL of water at 37 ± 0.5 0C. A standard stainless steel dissolution paddle rotating at 50 rpm provided gentle agitation. The in vitro performance of the formulations is visually assessed using the following grading system:
Grade A: Rapidly forming (within 1 min) nano emulsion, having a clear or bluish appearance.
Grade B: Rapidly forming, slightly less clear emulsion, having a bluish white appearance.
Grade C: Fine milky emulsion that formed within 2 min.
Grade D: Dull, grayish white emulsion having slightly oily appearance that is slow to emulsify (longer than 2 min).
Grade E: Formulation, exhibiting either poor or minimal emulsification with large oil globules present on the surface.
Grade A and Grade B formulation will remain as nanoemulsion when dispersed in GIT. While formulation falling in Grade C could be recommend for SEDDS formulation[73].
Turbidimetric Evaluation
Nepheloturbidimetric evaluation is done to monitor the growth of emulsification. Fixed quantity of Self-emulsifying system is added to fixed quantity of suitable medium (0.1N hydrochloric acid) under continuous stirring (50 rpm) on magnetic plate at ambient temperature, and the increase in turbidity is measured using a turbidimeter. However, since the time required for complete emulsification is too short, it is not possible to monitor the rate of change of turbidity (rate of emulsification) [44, 74]
Viscosity Determination
The SEDDS system is generally administered in soft gelatin or hard gelatin capsules. So, it can be easily pourable into capsules and such system should not too thick to create a problem. The rheological properties of the micro emulsion are evaluated by Brookfield viscometer. This viscosities determination conform whether the system is w/o or o/w. If system has low viscosity then it is o/w type of the system and if a high viscosity then it is w/o type of the system [44, 74].
Droplet Size Analysis Particle Size Measurements
The droplet size of the emulsions is determined by photon correlation spectroscopy (which analyses the fluctuations in light scattering due to Brownian motion of the particles) using a Zetasizer able to measure sizes between 10 and 5000 nm. Light scattering is monitored at 25°C at a 90° angle, after external standardization with spherical polystyrene beads. The nanometric size range of the particle is retained even after 100 times dilution with water which proves the system’s compatibility with excess water [44, 74].
Refractive Index and Percent Transmittance
Refractive index and percent tranmittance proved the transparency of formulation. The refractive index of the system is measured by refractometer by placing drop of solution on slide and it compare with water (1.333). The percent transmittance of the system is measured at particular wavelength using UV-spectrophotometer keeping distilled water as blank.If refractive index of system is similar to the refractive index of water(1.333) and formulation have percent transmittance > 99 percent, then formulation have transparent nature.
Electro conductivity Study
The SEDD system contains ionoc or non-ionic surfactant, oil, and water.so, this test is used to measure the electoconductive nature of system. The electro conductivity of resultant system is measured by electoconductometer.
In Vitro Diffusion Study
In vitro diffusion studies is performed to study the release behavior of formulation from liquid crystalline phase around the droplet using dialysis technique.[44]
Drug content
Drug from pre-weighed SEDDS is extracted by dissolving in suitable solvent. Drug content in the solvent extract was analyzed by suitable analytical method against the standard solvent solution of drug.
Limitations
One of the hindrances for the development of self-emulsifying drug delivery systems (SEDDS) and other lipid-based formulations is the lack of good predicative in vitro models for assessment of the formulations.
Traditional dissolution methods do not work, because these formulations potentially are dependent on digestion prior to release of the drug. To mimic this, an in vitro model simulating the digestive processes of the duodenum has been developed. This in vitro model needs further development and validation before its strength can be evaluated. Further development will be based on in vitro - in vivo correlations and therefore different prototype lipid based formulations needs to be developed and tested in vivo in a suitable animal model. Future studies will address the development of the in vitro model.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
APPLICATION
Improvement in Solubility and bioavailability:
If drug is incorporated in SEDDS, it increases the solubility because it circumvents the dissolution step in case of Class-? drug (Low solubility/high permeability). Ketoprofen, a moderately hydrophobic (log P 0.979) non-steroidal anti-inflammatory drug (NSAID), is a drug of choice for sustained release formulation has high potential for gastric irritation during chronic therapy. Also because of its low solubility, ketoprofen shows incomplete release from sustained release formulations. Vergote et al.(2001) reported complete drug release from sustained release formulations containing ketoprofen in nanocrystalline form [75] Different formulation approaches that have been sought to achieve sustained release, increase the bioavailability, and decrease the gastric irritation of ketoprofen include preparation of matrix pellets of nano-crystalline ketoprofen,70 sustained release ketoprofen microparticles [76] and formulations [76], floating oral ketoprofen systems [77], and transdermal systems of ketoprofen[78].
Preparation and stabilization of nano-crystalline or improved solubility forms of drug may pose processing, stability, and economic problems. This problem can be successfully overcome when Ketoprofen is presented in SEDDS formulation. This formulation enhanced bioavailability due to increase the solubility of drug and minimizes the gastric irritation. Also incorporation of gelling agent in SEDDS sustained the release of Ketoprofen. In SEDDS, the lipid matrix interacts readily with water, forming a fine particulate oil-in-water (o/w) emulsion. The emulsion droplets will deliver the drug to the gastro-intestinal mucosa in the dissolved state readily accessible for absorption. Therefore, increase in AUC i.e. bioavailability and Cmax is observed with many drugs when presented in SEDDS. These drugs are listed in table 2 & 3.
Table 2 Relative bioavailability of lipid based formulation of hydrophobic drugs
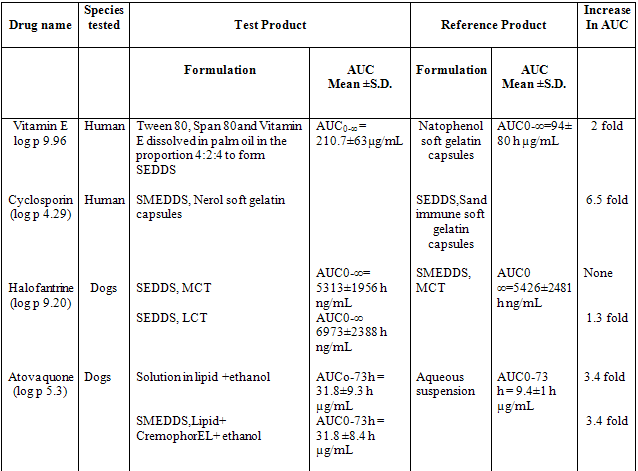
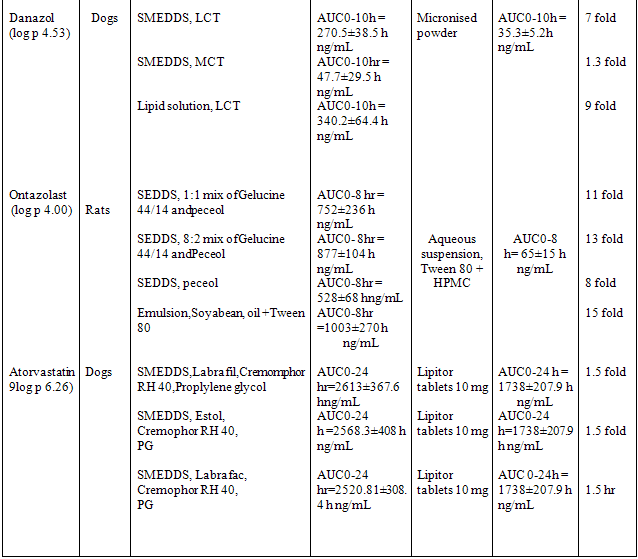
Table 3 Example of bioavailability enhancement of pooly soluble drug after administration of SEDDS and SMEDDS formulations
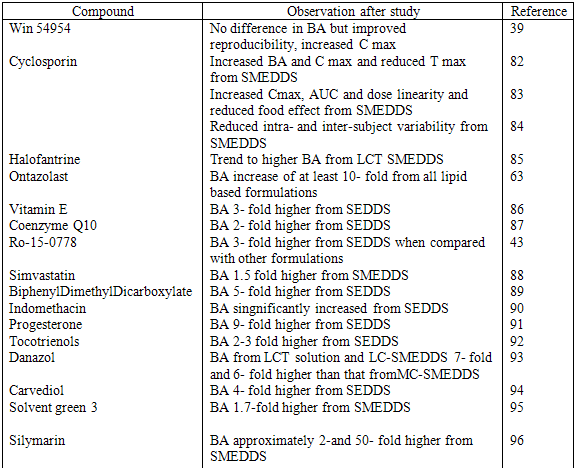

Protection against Biodegradation
The ability of self emulsifying drug delivery system to reduce degradation as well as improve absorption may be especially useful for drugs, for which both low solubility and degradation in the GI tract contribute to a low oral bioavailability. Many drugs are degraded in physiological system, may be because ofacidic PH in stomach, enzymatic degradation or hydrolytic degradation etc. Such drugs when presented in the form of SEDDS can be well protected against these degradation processes as liquid crystalline phase in SEDDS might be an act as barrier between degradating environment and the drug.
Acetylsalicylic acid (Log P = 1.2, Mw=180), a drug that degrades in the GI tract because it is readily hydrolyzed to salicylic acid in an acid environment. When the drug was formulated in a Galacticles™ Oral Lipid Matrix System (SEDDS formulation) and compare with a commercial formulation, it showed the good plasma profile as compare to reference formulation. The oral bioavailability of undegraded acetylsalicylic acid is improved by 73% by the Galacticles™ Oral Lipid Matrix System formulation compared to the reference formulation. This suggests that the SEDDS formulation has a capacity to protect drugs from degradation in the GI tract 43 Supersaturable SEDDS contain a reduced amount of a surfactant and a water-soluble cellulosic polymer (or other polymers) to prevent precipitation of the drug by generating and maintaining a supersaturated state in vivo.
The S-SEDDS formulations can result in enhanced oral absorption as compared with the related self-emulsifying drug delivery systems (SEDDS) formulation and the reduced surfactant levels may minimise gastrointestinal surfactant side effects.
Oral drug delivery systems are designed address the varied challenges in oral delivery of numerous promising compounds including poor aqueous solubility, poor absorption, and large molecular size. These are both liquid and powder-in-capsule products comprising our self-emulsifying liquid crystalline nano-particles (LCNP) technology (featuring Cubosome®, Hexosome®, and Flexosome™).
Liquid crystalline nano-particles (LCNPs) are excellent solubilizers. Compared with conventional lipid or non-lipid carriers, LCNPs show high drug carrier capacity for a range of sparingly water-soluble drugs. For drugs susceptible toin vivo degradation, such as peptides and proteins, LCNP vehicles protect the sensitive drug from enzymatic degradation. The LCNP systems also address permeability limitations by exploiting the lipid-mediated absorption mechanism. For water-soluble peptides typical bioavailability enhancements range from twenty to more than one hundred times. In an alternative application large proteins have been encapsulated for local activity in the gastrointestinal tract.
LCNP carriers can be combined with controlled-release and targeting functionalities. The particles are designed to form in situ at a controlled rate, which enables an effective in vivo distribution of the drug. LCNP carriers can also be released at different absorption sites, for example in the upper or lower intestine, which is important for drugs that have narrow regional absorption windows. SMEDDS” composition of PNU156804 that showed a good chemical stability and a higher bioavailability with respect to a conventional formulation.75
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Future Aspect
In relation to formulation development of poorly soluble drugs in the future, there are now techniques being used to convert liquid/semi-solid SEDDS and SMEDDS formulations into powders and granules, which can then be further processed into conventional 'powder-fill' capsules or even compressed into tablets. Hot melt granulation is a technique for producing granules or pellets, and by using a waxy solubilising agent as a binding agent, up to 25% solubilising agent can be incorporated in a formulation. There is also increasing interest in using inert adsorbents, such as the Neusilin (Fuji Chemicals) and Zeopharm (Huber) products for converting liquids into powders – which can then be processed into powder fill capsules or tablets. But to obtain solids with suitable processing properties, the ratio of SEDDS to solidifying excipients must be very high76, which seems to be practically non-feasible for drugs having limited solubility in oil phase. In this regard, it was hypothesized that the amount of solidifying excipients required for transformation of SEDDS in solid dosage forms will be significantly reduced if SEDDS is gelled. Colloidal silicon dioxide (Aerosil 200) is selected as a gelling agent for the oil based systems, which may serve the dual purpose of reducing the amount of solidifying excipients required and aiding in slowing drug release.
Reference:
1. G.L. Amidon, H et al., A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12
2. B.J. Aungst, Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism. J. Pharm. Sci. 1993; 82: 979-986.
3. D.L. Burcham, et al. Improved oral bioavailability of the hypocholesterolemic DMP 565 in dogs following oral dosing in oil and glycol solutions, Biopharm. Drug Dispos. 1997; 18: 737-742.
4. A.T.M. Serajuddin, et al. Effect of vehicle amphiphilicity on the dissolution and bioavailability of a poorly water-soluble drug from solid dispersion. J. Pharm. Sci. 1988; 77: 414-417.
5. B.J. Aungst, N. Nguyen, N.J. Rogers, S. Rowe, M. H Improved oral bioavailability of an HIV protease inhibitor using Gelucire 44/14 and Labrasol vehicles. B.T. Gattefosse 1994;87: 49-54.
6. M.G. Wakerly, C.W.Pouton, B.J. Meakin, F.S. Morton, Self-emulsification of vegetable oil-non-ionic surfactant mixtures. ACS Symp. Ser. 1986; 311: 242-255.
7. D.Q.M. Craig, et al. An investigation into the physico-chemical properties of self-emulsifying systems using low frequency dielectric spectroscopy, surface tension measurements and particle size analysis. Int. J. Pharm. 1993; 96: 147- 155.
8. H. Toguchi, Y. Ogawa, K. Iga,T. Yashiki, T. Shimamoto, Gastrointestinal absorption of ethyl 2 chloro-3-(4-(2-methyl-2-phenylpropyloxy) phenyl)propionate from different dosage forms in rats and dogs, Chem. Pharm. Bull. 1990; 38: 2792-2796.
9. T.T. Kararli, et al. Oral delivery of a renin inhibitor compound using emulsion formulations. Pharm. Res. 1992; 9: 888-893.
10. R.A. Schwendener,H. Schott, Lipophilic 1-beta-D-arabinofuranosyl cytosine derivatives in liposomal formulations for oral and parenteral antileukemic therapy in the murine L1210 leukemia model, J. Cancer Res. Clin. Oncol. 1996; 122: 723-726.
11. Kreuter J, Colloidal drug delivery systems.In: J,Kreuter editor. New York: Marcel Dekker, 1994.
12. Gregoriadis G. Liposome Technology.1993 2ndedition vol.1
13. Uchegbu IF, Florence AT. Non ionic surfactant vesicles(niosomes)physical and pharmaceutical chemistry.Adv Coll Int Sci 1995;58:1-55
14. Paul BK, Moulik SP. Microemulsion:An overview. Disp Sci . 1997; 18:301-367.
15. Moulik SP, Paul BK. Structure dynamics and transport properties of microemulsion.Adv Coll Int Sci 1998;78:99-199
16. Tenjarala S. Microemulsion: An overview and pharmaceuticalapplication .Crit Rev Ther drug carrier syst 1999;16:461-521
17. Kantarias,Rees GD,Lawrence MJ, Gelatin based organogels:rheology and application in ionophoretic transdermal drug delivery. J Contr Del. 1999; 60:355-365.
18. Watnasirichikul SS, et al. Preparation of biodegradeble insulin nanocapsules from biocompatible microemulsions. Pharm Res. 2002; 17:684-689
19. Lawrence MJ. Surfactant Systems: Their use in drug delivery. Chem. Soc Rev. 1994; 23:417-424
20. Cortesi R, Nastruzzi C. Liposomes, micelles and microemulsions as a new delivery system for cytotoxic alkaloids. Pharm Sci and Tech Today. 1995;2:288-289
21. Kahlweit M, Busse G,Faulhaber B..Preparing nontoxic microemulsions with alkyl monoglucosides and the role of alkane diols as cosolvent .Langmuir.1996; 12:861-862
22. Trotta M, Ugazio E, Gggasco MR.Psedoternary phase diagram of lecithin based microemusion:influence of monoalkly phosphate. J Phar Pharmacol. 1995; 47:451-454
23. Das ML,Bhattacharya PK, Moulik SP, Model biological microemulsoion :Part1. Phase behavior and physicochemical properties of cholesteryl benzoate and sodium deoxycholate contained microemulsion Ind J Biochem Biophys .1989; 26:24-29
24. Mukhopadhyay L, et al.Thermodyanamics of formation of biological microemulsion with cinnamic alcohol,AOT, tween-20 and water and kinetics of alkaline fading of crastal violet inthem. J Coll Int Sci. 1997; 186: 1-8
25. Acharya A, et al.Physicochemical investigation of microemulsification of coconut oil and water using polyoxyethylene 2-cetyl ether(brij-52) and isopropanol or ethanol. J Coll Int Sci. 2002; 245:163-170
26. Majhi P, Moulik SP, Physicochemical studies on biological macro and microemulsion.vi.mixing behavior of eucalyptus oil, water and polyoxyethylene sorbitan monolaurate (tween -20)assisted by n-butanol and cinnamic alcohol. J Disp Sci Tech. 1999; 20:1407-1427
27. Gupta S, et al. Preparation of prospective plant oil derived microemulsion vehicles for drug delivery. Ind J Biochem Biophys. 2006; 43:254-257
28. Shukla A, et al. Microemulsion for drug delivery studied by dynamic light scattering:effect of interparticle intreractions in oil-in-water microemulsion . J Pharm Sci. 2001; 92:730-738
29. Kreilgaard M, Pederson EJ, Jarozewski JW, NMR characterization and transdermal drug delivery potential of microemulsion systems. J control Rel. 2000; 69:421-433
30. Malcomson C, Lawrence MJ. Comparison of the incorporation of model steroids into non-ionocmicellar and microemulsion systems. J Pharm Pharmacol. 1993; 45:141-143
31.Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption :physical and biopharmaceutical aspects. Pharm Res . 1995; 12:1561-1572
32. Gupta S, et al. Designing and testing of an effective oil-in-water microemulsion drug delivery system for in vivo application. Drug Del. 2005;12:267-273
33. Kagan A garti N, Microemulsions as transdermal drug delivery vehicles. Adv coll int sci. 2006:123-126:369-385
34. Hazra B, et al.Microemulsion encapsulation of diospyrin a plant-derived bisnapthoquinonoid of potential chemotherapeutics activity. J Pharm Pharmacol. 1998; 50:191-196
35. Von Corswant C, et al. Microemulsions based on soyaben phosphatidylcholine and triglycerides phase behavior and microstructure. Langmuir. 1997;13:507-561
36.Trotta M, Gasco MR, Morel S, .Release of drugs from oil-water microemulsion. J Contr Rel. 1989; 10:237-243
37. Von Corswant C ,Thorne P, Engstrom S. Triglycerides based microemulsion for intravenous administration of sparingly soluble substances J Pharm Sci. 1998: 87:200-208
38. Moreno MA, et al. Lyophilised lecithin based oil-in-water microemulsion as new and low toxic delivery system for amphotericin B .Pharm Res. 2001; 18:344-351
39. Charman SA, et al. Self emulsifying drug delivery systems: formulation and biopharmaceutical evaluation of an investigational lipophilic compound. Pharm Res. 1992; 9:87-93.
40. P.P. Constantinides, Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects, Pharm. Res. 1995; 12: 1561-1572.
41. D.Q.M. Craig, The use of self-emulsifying systems as a means of improving drug delivery, B.T. Gattefosse 1993; 86; 21-31.
42. Pouton CW. SEDDS: Assessment of the efficiency of emulsification.Int J Pharm. 1985; 27:335-348.
43. Shah NH, et al. Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm. 1994; 106:15-23.
44. Patil P, Joshij, paradkar. Effect of formulatiuon variables on preparation and evaluation of gelled self-emulsifying drug delivery system(SEDDS)of ketoprofen.AAPS Pharm Sci Tech.2004;5(3):34-42
45. Pouton CW, Charman WN. The potential of oily formulations for drug delivery to the gastro-intestinal tract. Adv Drug Deliv Rev. 1997; 25:1-2.
46. A.T.M. Serajuddin, et al. Effect of vehicle amphiphilicity on the dissolution and bioavailability of a poorly water-soluble drug from solid dispersion, J. Pharm. Sci. 1988; 77: 414-417.
47. Mittal pooja et al. , Potential assessment of Transcutol P and Lauroglycol FCC as Co-Surfactants forformulation of self Microemulsifying Drug Delivery Systems (Smedds), International Journal Pharmaceutical Sciences;2012:4(1),1742-1745
48. Shinde Ganesh et al: Self Microemulsifying Drug Delivery System: A Novel approach for Hydrophobic Drugs,International Journal Pharmaceutical Sciences,Jan-April: 2011;3(1)
49.KHAMKAR GANESH S,SELF MICRO EMULSIFYING DRUG DELIVERY SYSTEM (SMEED) O/W MICROEMULSION FOR BCS CLASS II DRUGS: A APPROACH TO ENHANCE AN ORAL BIOAVAILABILITY;Int. J. Pharm. Pharm. Sci,2011,3(3),13.
50.RAJAN B MISTRY, NIRAV S SHETH;A REVIEW: SELF EMULSIFYING DRUG DELIVERY SYSTEM; Int J Pharm Pharm Sci, 2011,3(2),2328.
51. Sachan R. etal, Self-Eumlsifying Drug Delivery System A Novel Approach for enhancement of Bioavalibility, Int.J. PharmTech Res.;2010,2(3):1738-1745.
52. Amidon, G.., et.al.A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability, Pharm.Res. 1995; 12: 413-420.
53. H. Reiss, Entropy-induced dispersion of bulk liquids, J. Colloids Interface Sci. 1975; 53:61-70.
54. T. Dabros, et al. Emulsification through area contraction, J. Colloids Interface Sci.1999; 210 :222-230
55. M.J. Groves, R.M.A. Mustafa, J.E. Carless, Phase studies of mixed phosphated surfactantsn-hexane and water, J. Pharm. Pharmacol. 1974; 26: 616-623.
56. M.J. Rang, C.A. Miller, Spontaneous emulsification of oils containing hydrocarbon, non-ionic surfactant, and oleyl alcohol, J. Colloids Interface Sci. 1999; 209: 179-192.
57. M.G. Wakerly, C.W.Pouton, B.J. Meakin, Evaluation of the selfemulsifying performance of a non-ionic surfactant-vegetable oil mixture, J. Pharm. Pharmacol. 1987; 39: 6P.
58. C.W. Pouton, M.G. Wakerly, B.J. Meakin, Self-emulsifying systems for oral delivery of drugs, Proc. Int. Symp. Control. Release Bioact. Mater. 1987; 14: 113-114.
59. D.Q.M. Craig, et al. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy,Int. J. Pharm. 1995; 114: 103-110.
60. C.W. Pouton, Self-emulsifying drug delivery systems: assessment of the efficiency of emulsification, Int. J. Pharm. 1985; 27: 335-348.
61. Grove M., et al. Bioavailability of seocalcitol II: development and characterisation of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglycerides . Eur J Pharm Sci. 2004 ; 28: 233-242.
62. M. Kimura, et al. Relationship between the molecular structures and emulsification properties of edible oils, Biosci. Biotech. Biochem. 1994;58: 1258-1261.
63. D.J. Hauss, et al. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor, J. Pharm. Sci. 1998; 87: 164-169.
64. N. Farah, J.P. Laforet, J. Denis, Self-microemulsifying drug delivery systems for improving dissolution of drugs: in vitro/in vivo evaluation, Pharm. Res. ; 11:1994 S202.
65. H. Yuasa, et al Evaluation of milk fatglobule membrane (MFGM) emulsion for oral administration: absorption of a-linolenic acid in rats and the effect of emulsion droplet size, Biol. Pharm. Bull. 1994; 17:756-758.
66. E. Georgakopoulos, N. Farah, G. Vergnault, Oral anhydrous nonionic microemulsions administrated in softgel capsules, B.T. Gattefosse 1992; 85:11-20.
67. E.S. Swenson, W.B. Milisen, W. Curatolo, Intestinal permeability enhancement: efficacy, acute local toxicity and reversibility, Pharm. Res. 1994; 11:1132-1142.
68. T. Amemiya, S. Mizuno, H. Yuasa,J. Watanabe, Development of emulsion type new vehicle for soft gelatin capsule. I. Selection of surfactants for development of new vehicle and its physicochemical properties, Chem. Pharm. Bull. 1998; 47: 309-313.
69. A. Meinzer, E. Mueller, J. Vonderscher, Microemulsion a suitable galenical approach for the absorption enhancement of low soluble compounds. B.T. Gattefosse 1995; 88: 21-26.
70. J. Vonderscher, A. Meinzer, Rationale for the development of Sandimmune Neoral. Transplant. Proc. 1994; 26: 2925-2927.
71. A. Karim, et al. HIV protease inhibitor SC-52151: a novel method of optimizing bioavailability profile via a microemulsion drug delivery system, Pharm. Res. 1994; 11:S368.
72. I.F. Uchegbu, S.P. Vyas, Non-ionic surfactant based vesicles (niosomes) in drug delivery, Int. J. Pharm. 1998; 172: 33-70.
73. S. Shafiq, et al. Development and bioavailability assessment of ramipril nanoemulsion formulation Eur. J. Pharm. Biopharm 2007; 66: 227–243.
74. patil p, Vandana p, paradkar p formulation of self-emulsifying drug delivery system for oraldelivery of simvastatin:In vitro and in vivo evaluation. Acta pharma. 2007; 57: 111-122.
75. Vergote GJ, et al. An oral controlled release matrix pellet formulation containing nanocrystalline ketoprofen. Int J Pharm. 2001; 219:81-87.
76. Yamada T, Onishi H, Machida Y. Sustained release ketoprofen microparticles with ethylcellulose and carboxymethylethylcellulose. J Control Release. 2001;75:271-282.
77. Roda A, et al. Bioavailability of a new ketoprofen formulation for once-daily oral administration. Int J Pharm. 2002;241:165-172.
78. El-Kamel AH, et al. Preparation and evaluation of ketoprofen floating oral delivery system. Int J Pharm. 2001;220:13-21.
79. Rhee Y-S, et al. Transdermal delivery of ketoprofen using microemulsions. Int J Pharm. 2001; 228:161-170.
80. P.Gao, et al. Enhanced oral bioavailability of a poorly water soluble drug PNU- 91325 by supersaturable formulations, Drug Dev.Ind.Pharm.2004;30:221-229.
81. Schwarz J. Solid self-emulsifying dosage form for improved delivery of poorly soluble hydrophobic compounds and the process of preparation thereof. US patent application No. 2003 0 072 789. 2003.
82. A.K. Trull, et al. Enhanced absorption of new oral cyclosporin microemulsion formulation, Neoral, in liver transplant recipients with external biliary diversion, Transplant. Proc. 1994; 26: 2977–2978.
83. E.A. Mueller, et al. Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion formulation, Pharm. Res. 1994;11: 301–304.
84. J.M. Kovarik, et al Reduced inter- and intraindividual variability in cyclosporine pharmacokinetics from a microemulsion formulation, J. Pharm. Sci 1994; 83: 444–446.
85. S.-M. Khoo, et al. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of Halofantrine, Int. J. Pharm. 1998;167: 155–164.
86. T. Julianto, K.H. Yuen, A. Mohammad Noor, Improved bioavailability of vitamin E with a self emulsifying formulation, Int. J. Pharm. 2000; 200: 53–57.
87. T.R. Kommuru, et al. Self-emulsifying drug delivery systems (SEDDS) of coenzymeQ10: formulation development and bioavailability assessment, Int. J. Pharm. 2001;212: 233–246.
88. B.K. Kang, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin i








