
{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
Vikash Kumar Chaudhari1*, Vijay Yadav2, Praveen Kumar Verma1, Amit Kumar Singh2
1Department of Pharmaceutical Chemistry,
2Department of Pharmacognosy,
Kunwar Haribansh Singh College of Pharmacy, Jaunpur, U.P.
*vikashk464@gmail.com
ABSTRACT
Good manufacturing practices (GMP) is a part of quality assurance which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization. GMP guidelines provide minimum requirements for pharmaceutical or a food product manufacturer must meet to assure that the products are of high quality and do not pose any risk to the consumer or public.
INTRODUCTION
The term GMP was introduced to regulate manufacturing and packaging operations in the pharmaceutical industry. The Medicine Inspector of the Department of Health and Social Security of England, in consultation with other interested bodies compiled the guide to GMP also known as the Orange Guide. The first edition of the guide was published in 1971, the manufacturing of drug carried out under the Medicines Act. It was a relatively light volume of 20 pages, and was reissue third impression in 1972, with the addition of a 2-page appendix on sterile medicinal products. The color of its cover, it known as the Orange Guide. The second edition (52 pages, including five appendices) was published in 1977. The third edition (110 pages, five appendices) was published in 1983[1].
The Medicines and Healthcare products Regulatory Agency (MHRA) has published new edition of the Orange Guide in 2007. In United States, the first GMP regulations were issued in 1963 and described the GMP to be followed in the manufacture, packaging, and storage of finished pharmaceutical products. GMP regulations were developed by the US FDA and issued the United States CFR Chapter 21 in 1978. The regulation was similar in concept to the Orange Guide, but enforceable by law whereas the UK guide as an advisory. US congress passed the Federal Ani-tempering Act in 1983, making it a crime to tamper with packaged consumer products [2].
In the 1980, US FDA began publishing series of guidance documents that have a major effect on our interpretation of current GMP (cGMP). A “Guide to Inspection of Computerized Systems in Drug Processing” was published in 1983 and “Guideline on General Principles of Process Validation” was published in 1987. March 1997, the US FDA issued 21 CFR Part 11 which dealt with the use of electronic records and signatures. In 2000, US FDA introduced a guidance document on the incorporation of risk management into device development [3].
GOOD MANUFACTURING PRACTICES (GMP) GUIDELINES
Many countries have legislated that pharmaceutical and medical device companies created their own GMP guidelines that correspond with their legislation. Basic concepts of GMP guidelines goal of safeguarding the health of the patient as well as producing good quality medicine, medical devices or active pharmaceutical products[4]. The formalization of GMP commenced in the 1960s and their effect in over 100 countries ranging from Afghanistan to Zimbabwe. Examples of these include the following.
a. Pharmaceutical Inspection Convention (PIC):- Guide to GMP for pharmaceutical products-Australia, Austria, Belgium, Canada, Italy, Latvia, Liechtenstein, Denmark, Finland, France, Hungary, Ireland, Malaysia, The Netherlands, Norway, Poland, Portugal, Romania, Singapore, Slovak Republic, Spain, Sweden, Switzerland, and the United Kingdom.
b. Association of South-East Asia Nations (ASEAN):- General guidelines Brunei Darussalaam, Indonesia, Lao PDR, Malaysia,Cambodia, Myanmar, Philippines, Singapore, Thailand, and Vietnam.
c. European Economic Community (EEC):- Guide to GMP for medicinal products Austria, Belgium, Denmark, Ireland, Italy, Luxembourg, the Netherlands, Finland, France, Germany, Greece, Portugal, Spain, Sweden, and the United Kingdom.
In general, GMP has been issued guides to the achievement of consistent product quality, with interpretation and individual variations being accepted. GMP enforced in the United States by the US FDA, under Section 501(B) of the 1938 Food, Drug, and Cosmetic Act (21 USCS § 351). The regulations use the phrase "current good manufacturing practices" (cGMP) and it describes the guidelines [5].
The World Health Organization (WHO) version of GMP is used by pharmaceutical regulators and the pharmaceutical industry in over one hundred countries worldwide, primarily in the developing world including country like Nepal. The European Union's GMP (EU-GMP) enforces similar requirements to WHO GMP, as does the Food and Drug Administration's version in the US. Similar GMPs are used in other countries, with Australia, Japan, Canada, Singapore and others having highly developed/sophisticated GMP requirements. In the United Kingdom, the Medicines Act (1968) covers most aspects of GMP commonly referred to as "The Orange Guide", which is officially known as Rules and Guidance for Pharmaceutical Manufacturers and Distributors [6].
GMP inspections are performed in the United Kingdom by the Medicines and Healthcare Products Regulatory Agency (MHRA); in the Republic of Korea (South Korea) by the Korea Food & Drug Administration (KFDA); in Australia by the Therapeutically Goods Administration (TGA); in South Africa by the Medicines Control Council (MCC); in Brazil by the Agencia Nacional de Vigilancia Sanitaria (National Health Surveillance Agency Brazil) (ANVISA). In India GMP inspections are carried out by state Food and Drug Administration (FDA) and these FDA report to Central Drugs Standard Control Organization: in Nepal, GMP inspections are carried out by the Department of Drug Administration (DDA) and in Pakistan by the Ministry of Health. Nigeria has National Agency for Food and Drug Administration and Control (NAFDAC) [7].
COMPONENTS OF GMP
GMP requires that the manufacturing process is fully defined before being initiated and all the necessary facilities are provided. In practice, personnel must be adequately trained, suitable premises and equipment used, correct materials used, approved procedures adopted, suitable storage and transport facilities available, and appropriate records made[8]. The essential components of GMP are summarized in Figure 1.
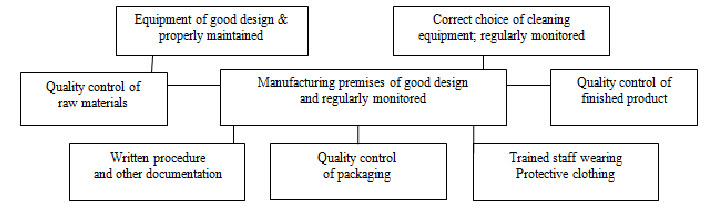
Fig.1. Components of Good Manufacturing Practice
Indian schedule M for GMP and requirements of premises, plant and equipment for pharmaceutical products. Part I includes general requirements, Warehousing area, Production area, Quality control area, Personnel, Ancillary area, Health, clothing and sanitation of workers, Manufacturing operations and controls, Sanitation in the manufacturing premises, Raw materials, Equipment, Documentation and Records, Labels and other printed materials, Quality assurance, Self inspection and quality audit, Quality control system, Specification, Master formula records, Packing records, Batch packaging records, Batch processing records, Standard operating procedures (SOPs) and records, Reference samples, Reprocessing and recoveries, Distribution records, Validation and process validation, Product recalls, Complaints and adverse reactions and Site-master file. Part I-A to part I-E mentions about the specific requirements for manufacture of different products and Part I-F mentions about the specific requirements of premises, plant and materials for manufacture of active pharmaceutical ingredients (bulk drugs). Part II describes the Requirement of plant and equipments for various dosage forms [4].
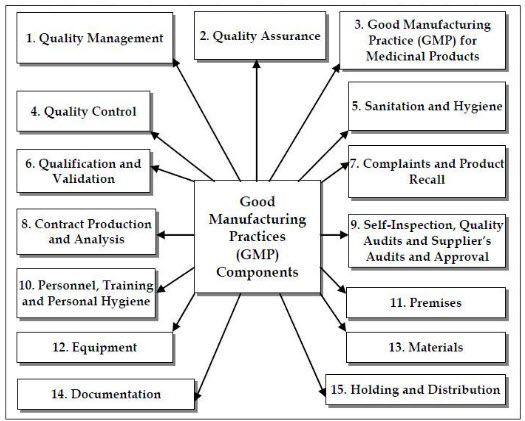
Fig. 2. Consolidated Components of Good Manufacturing Practices
QUALITY MANAGEMENT
The holder of a manufacturing authorization must manufacture medicinal products so as to ensure that they are fit for their intended use, comply with the requirements of the marketing authorization and do not place patients at risk due to inadequate safety, quality or efficacy. The attainment of this quality objective is the responsibility of senior management and requires the participation and commitment by staff in many different departments and at all levels within the company, by the company’s suppliers and by the distributors [9].
In the pharmaceutical industry at large, quality management is usually defined as the aspect of management function that determines and implements the “quality policy”, i.e. the overall intention and direction of an organization regarding quality, as formally expressed and authorized by top management[10].
QUALITY ASSURANCE (QA)
QA is a wide ranging concept, which covers all matters, which individually or collectively influence the quality of a product. It is the sum total of the organized arrangements made with the objective of ensuring that pharmaceutical products are of the quality required for their intended use. QA, therefore, incorporates GMP and other factors such as product design and development [11].
The system of QA appropriate for the manufacture of pharmaceutical products should ensure that:
a. Pharmaceutical products are designed and developed in a way that takes account of the requirements of GMP and other associated codes such as those of good laboratory practice (GLP) and good clinical practice (GCP).
b. Production and control operations are clearly specified in a written form and GMP requirements are adopted.
c. Arrangements are made for the manufacture, supply and use of the correct starting and packaging materials.
d. All necessary controls on starting materials, intermediate products, and bulk products and other in-process controls, calibrations, and validations are carried out.
e. The finished product is correctly processed and checked, according to the defined procedures, pharmaceutical products are not sold or supplied before the authorized persons have certified that each production batch has been produced and controlled in accordance with the requirements of the marketing authorization and any other regulations relevant to the production, control and release of pharmaceutical products.
f. Satisfactory arrangements exist to ensure, as far as possible, that the pharmaceutical products are stored by the manufacturer, distributed, and subsequently handled so that quality is maintained throughout their shelf-life;
g. Deviations are reported, investigated and recorded.
h. Regular evaluations of the quality of pharmaceutical products should be conducted with the objective of verifying the consistency of the process and ensuring its continuous improvement.
GOOD MANUFACTURING PRACTICE (GMP) FOR MEDICINAL PRODUCTS
GMP is a part of QA which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorizations or product specification. GMP is aimed primarily at diminishing the risks inherent in any pharmaceutical production [12].
The basic requirements of GMP are that:
a. All manufacturing processes are clearly defined, systematically reviewed in the light of experience, and shown to be capable of consistently manufacturing pharmaceutical products of the required quality that comply with their specifications.
b. Qualification and validation are performed.
c. All necessary resources are provided.
d. Instructions and procedures are written in clear and unambiguous language, specifically applicable to the facilities provided.
e. Operators are trained to carry out procedures correctly.
f. Records are made during manufacture to show that all the steps required by the defined procedures and instructions have in fact been taken and that the quantity and quality of the product are as expected; any significant deviations are fully recorded and investigated.
g. Records manufacture and distribution, which enable the complete history of a batch to be traced, are retained in a comprehensible and accessible form.
h. The proper storage and distribution of the products minimizes any risk to their quality.
i. A system is available to recall any batch of product from sale or supply.
j. Complaints about marketed products are examined the causes of quality defects investigated, and appropriate measures taken in respect of the defective products to prevent recurrence [13].
QUALITY CONTROL (QC)
QC is a part of GMP which is concerned with sampling, specifications and testing, and with the organization, documentation and release procedures which ensure that the necessary and relevant tests are actually carried out and that materials are not released for use, nor products released for sale or supply, until their quality has been judged to be satisfactory. QC is not confined to laboratory operations, but may be involved in many decisions concerning the quality of the product. QC establish, validate and implement all QC procedures, to evaluate, maintain, and store the reference standards for substances, to ensure the correct labeling of containers of materials and products, to ensure that the stability of the active pharmaceutical ingredients (APIs) and products is monitored, to participate in the investigation of complaints related to the quality of the product, and to participate in environmental monitoring[14].
SANITATION AND HYGIENE
A high level of sanitation and hygiene should be practiced in every aspect of the manufacture of medicine products. The scope of sanitation and hygiene covers personnel, premises, equipment and apparatus, production materials and containers, products for cleaning and disinfection, and anything that could become a source of contamination to the product. Potential sources of contamination should be eliminated through an integrated comprehensive programme of sanitation and hygiene[15].
The areas, surfaces, and equipment in and on which products are made must be kept clean. Dirt, and the microbes that it can harbor, must not get into or on products. Disinfectants can be inactivated by dirt. Dirt (particularly oily or greasy films and protein like matter) can also protect microorganisms against the action of disinfectants. So, before disinfection, it is important to first clean surfaces. Where gross amounts of dirt are present, it may be necessary to first remove most of it by scrubbing. Then surfaces may be cleaned by the application of a cleaning agent, followed by rinsing[16].
QUALIFICATION AND VALIDATION
Qualification is an action of providing that any premises, systems and items of equipment work correctly and actually lead to the expected results. Validation is defined as the establishing of documented evidence which provides a high degree of assurance that a planned process will consistently perform according to the intended specified outcomes. Validation studies should reinforce GMP and be conducted in accordance with defined procedures. Results and conclusions should be recorded. When any new manufacturing formula or method of preparation is adopted, steps should be taken to demonstrate its suitability for routine processing [17].
Qualification and validation should establish and provide documentary evidence that:
a. The premises, supporting utilities, equipment and processes have been designed in accordance with the requirements for GMP (Design qualification or DQ).
b. The premises, supporting utilities and equipment have been built and installed in compliance with their design specifications (Installation qualification or IQ).
c. The premises, supporting utilities and equipment operate in accordance with their design specifications (Operational qualification or OQ).
d. A specific process will consistently produce a product meeting its predetermined specifications and quality attributes (Process validation or PV, also called performance qualification or PQ).
COMPLAINTS AND PRODUCT RECALLS
QA and GMP are about preventing errors. However, in this imperfect universe there is no such thing as an infallibly perfect system, and an essential feature of any QA system is a plan for dealing with complaints, or reports of faulty products, if they do occur. A requirement to cover this occurs in all notable GMP guidelines. Complaints received from consumers, professionals and the trade serves as a primary means of obtaining feedback about product quality after distribution. It is necessary, therefore, that each complaint or inquiry be evaluated by knowledgeable and responsible personnel [3].
The records of packaging, production and distribution of drug and the retained samples provide the basis for assessing the validity and seriousness of the alleged deviations that precipitated the complaint. The complaint file itself also plays an important role in determining whether any other similar complaints have been received on the lot in question, or on any other lots of the same product. The evaluation of complaints serves several valuable purposes. First, there is the urgent need to confirm whether consumers are potentially at risk and to initiate any appropriate action. A second value is the review of the product and its production process to establish whether any modifications are required. Third is the need to rapidly respond to the customer, thereby attempting to maintain confidence in the product and company [18].
CONTRACT PRODUCTION AND ANALYSIS
The global industry is changing its shape through rationalization, mergers and acquisitions. Companies are increasingly considering the use of other manufacturers to produce or manufacture their products. Companies are also finding that they do not have the technology or expertise to manufacture certain new special dosage forms. In some cases, financial targets mean that companies are not using manufacturing as a core business process. This means that the importance of contract manufacturing and testing of products is also increasing [19].
The main principle underlying contract production and analysis is very simple. The work has to be clearly defined, agreed and controlled to avoid misunderstandings. The simplest way to avoid such misunderstandings is to have a written contract, setting out the duties of all parties to the contract and the standards that must be met.
SELF INSPECTION, QUALITY AUDITS AND SUPPLIER’SAUDITS/ APPROVAL
The purpose of self-inspection is to evaluate the manufacturer’s compliance with GMP in all aspects of production and quality control. The self-inspection programme should be designed to detect any shortcomings in the implementation of GMP and to recommend the necessary corrective actions. Self-inspections should be performed routinely, and may be, in addition, performed on special occasions, e.g. in the case of product recalls or repeated rejections, or when an inspection by the health authorities is announced. The team responsible for self-inspection should consist of personnel who can evaluate the implementation of GMP objectively. Management appoints a self-inspection team consisting of experts in their respective fields and familiar with GMP [17].
PERSONNEL, TRAINING AND PERSONAL HYGIENE
The establishment and maintenance of a satisfactory system of quality assurance and the correct manufacture and control of pharmaceutical products and active ingredients. For this reason there must be sufficient qualified personnel to carry out all the tasks for which the manufacturer is responsible. Individual responsibilities should be clearly defined and understood by the persons concerned and recorded as written job descriptions [10]. Personnel should be aware of the principles of GMP that affect them and receive initial and continuing training, including hygiene instructions, relevant to their need [16]. In order to effectively monitor and control virtually all GMP documents/activities in a facility, the quality professional should have a very high level of skills, knowledge and experience [3].
Peoples are the most important factor in the assurance of quality. This is true of all levels within an organization, from company president and managing director to the most-junior employee. It may well be possible for high-quality, well-trained, dedicated personnel to compensate for a lack or deficiency in the other elements. Nothing, not even the finest equipment, premises, materials, or procedures can compensate for the quality hazard represented by low-standard, ill-trained, or poorly motivated staff. Self-responsible, well-motivated staff will produce more goods, with a greater assurance of the quality of those goods, than will poorly motivated staff. Conversely, in the special context of medicines manufacture, poorly motivated staff can represent a hazard to themselves, to the public, and to company profits [20].
PREMISES
Premises must be located, constructed, adapted, designed, and maintained to suit the operations to be carried out. The layout and design of premises must aim to minimize the risk of errors and permit effective cleaning and maintenance in order to avoid cross contamination, build-up of dust or dirt, and in general, any adverse effect on the quality of products[20]. The choices of materials of construction for manufacturing facilities are numerous. Some examples are presented subsequently [21].
a. Walls: Walls in manufacturing areas, packaging areas and corridors should be of plaster finish on high-quality concrete blocks or gypsum board. The finish should be smooth, usually with enamel or epoxy paint. They should be washable and able to resist repeated applications of cleaning and disinfecting agents.
b. Floors: Floor covering should be selected for durability as well as for clean ability and resistance to the chemicals with which it is likely to come into contact. Epoxy flooring provides a durable and readily cleanable surface.
c. Ceilings: Suspended ceilings may be provided in office areas, toilets, laboratories and cafeterias. They usually consist of lay-in acoustical panels of non brittle, non friable, non asbestos and non combustible material. Manufacturing areas require a smooth finish, often of seamless plaster or gypsum board. All ceiling fixtures such as light fittings, air outlets and returns should be designed to assure ease of cleaning and to minimize the potential for accumulation of dust.
d. Services: In the building design, provisions must be made for drains, steam, electricity, water and other services to allow for ease of maintenance. Access should, ideally, be possible without disruption of activity within the actual rooms provided with the services. Doors and window-frames should all have a hard, smooth, impervious finish, and should close tightly. Window and door frames should be fitted flush, at least on sides facing inward to processing areas. Doors, except emergency exits, should not open directly from production areas to the outside world. Any emergency exit doors should be kept shut and sealed, and designed so as to be open able only when emergency demands [22].
EQUIPMENT
Manufacturing equipment should be capable of producing materials, products and intermediates that are intended and that conform to the required or specified quality characteristics. The equipment must be designed and built so that it is possible to clean it thoroughly. Surfaces that come into contact with products should have polished finishes, smooth with no recesses, crevices, difficult corners, uneven joints, dead-legs, projections, or rough welds to harbor contamination or make cleaning difficult. Equipment must also be capable of withstanding repeated, thorough cleaning. Traces of previous product, at levels that might be acceptable in other industries, are totally unacceptable in the manufacture of medicines. Between batches all manufacturing equipment must be thoroughly cleaned and disinfected or sterilized [23].
MATERIALS
A flow diagram illustrating the ordering, receipt, sampling, approval (or rejection), and dispensing of starting materials is shown in Figure 3. The purchasing department orders the material on the basis of a starting material specification provided to them by the QC.
Purchasing department sends the order to an approved supplier, which is a company that has been approved, jointly by the QA, QC and production to supply the material in question. Different suppliers batches within one delivery are to be segregated, one from another, with a different internal lot number for each entered on the QUARANTINE label, which is applied to each container. If all the containers in the delivery appear to be correct and in good condition, the goods inwards departments then place on each container a QUARANTINE label with the entries for “Code Number,” “Name of Material,” “Lot Number,” and “Date Received” are completed [17].
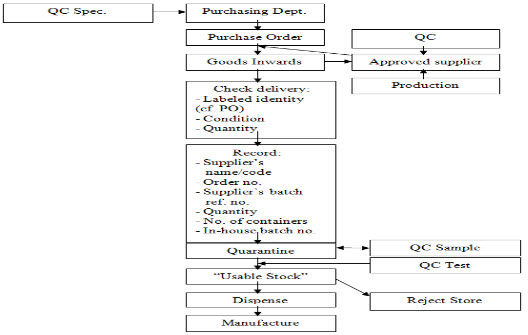
Fig. 3. Components/Starting Materials Flow Chart
Notes:
a. It is useful to have the QUARANTINE label and the RELEASED and REJECTED labels printed in different colors, for example, for QUARANTINE, black print on a yellow background, for RELEASED green print on a white background, and for REJECTED red print on a white background.
b. In the examples shown, the intention is that, when the QC decision is made, the RELEASED (or REJECTED) label should be applied just over the lower QUARANTINE panel. This may seem an infringement of the golden rule about not applying new labels over old ones, but here (if the, say, RELEASED label falls off, or is removed) the labeled status of the material reverts to QUARANTINE, i.e., it is fail-safe. The benefit of the labeling system illustrated is the elimination of any possible error in transcribing the information originally entered on the QUARANTINE label.
PACKAGING MATERIALS
The purchase, sampling, release, receipt and control of printed packaging materials and primary packaging materials (that is packaging materials that come into direct contact with the product, as compared with secondary packaging materials, which do not) need to be accorded the same level of attention as given to starting materials. Documents, records, and procedures analogous to those outlined above should be employed. All the regulatory requirements are in agreement that components or (starting materials) and containers, etc. should be storage is to “permit batch segregation and stock rotation” [FEFO, “first expiry, first out” or FIFO “first in, first out”]. It also permits wet-cleaning of floors, without the risk of wetting the materials [24].
DOCUMENTATION
Good documentation is an essential part of the QA system and GMP. The various types of documents used should be fully defined in the manufacturer's quality management system (QMS). The main objective of the system of documentation utilized must be to establish, control, monitor and record all activities which directly or indirectly impact on all aspects of the quality of medicinal products [9].
Given below is a list of the most common types of documents along with a brief description of each.
Site Master File: A document describing the GMP related activities of the manufacturer.
Quality Manual: A global company document that describes, in paragraph form, the regulations and/or parts of the regulations that the company is required to follow.
Policies: Documents that describe in general terms, and not with step-by-step instructions, how specific GMP aspects (such as security, documentation, health, and responsibilities) will be implemented.
Standard Operating Procedures (SOPs): Step-by-step instructions for performing operational tasks or activities.
Batch Records: These documents are typically used and completed by the manufacturing department. Batch records provide step-by-step instructions for production-related tasks and activities, besides including areas on the batch record itself for documenting such tasks.
Test Methods: These documents are typically used and completed by the quality control (QC) department. Test methods provide step-by-step instructions for testing supplies, materials, products, and other production-related tasks and activities, e.g., environmental monitoring of the GMP facility.
Logbooks: Logbooks are used for documenting the operation, maintenance, and calibration of a piece of equipment.
Logbooks are also used to record critical activities, e.g., monitoring of clean rooms, solution preparation, recording of deviation, change controls and its corrective action assignment.
Batch processing record- A batch processing record should be kept for each batch processed. It should be based on the relevant parts of the currently approved manufacturing formula and processing instructions, and should contain the following information:
a. The name and batch number of the product [9].
b. Dates and times of commencement, of significant intermediate stages and of completion of production.
c. Identification of the operator who performed each significant step of the process and, where appropriate, the name of any person who checked these operations.
d. The batch number and analytical control number as well as the quantities of each starting material actually weighed.
e. A record of the in-process controls and the initials of the person(s) carrying them out, and the results obtained.
f. The product yield obtained at different and pertinent stages of manufacture.
g. Notes on special problems including details, with signed authorization for any deviation from the manufacturing formula and processing instructions.
h. Approval by the person responsible for the processing operations.
Batch packaging record-A batch packaging record should be kept for each batch or part batch processed. The batch packaging record should contain the following information [13]:
a. The name and batch number of the product.
b. The date and times of the packaging operations.
c. Identification of the operator who performed each significant step of the process and, where appropriate, the name of any person who checked these operations.
d. Records of checks for identity and conformity with the packaging instructions, including the results of in-process controls.
e. Details of the packaging operations carried out, including references to equipment and the packaging lines used.
f. Whenever possible, samples of printed packaging materials used, including specimens of the batch coding, expiry dating and any additional overprinting.
g. The quantities and reference number or identification of all printed packaging materials and bulk product issued, used, destroyed or returned to stock and the quantities of obtained product, in order to provide for an adequate reconciliation.
h. Approval by the person responsible for the packaging operations.
HOLDING AND DISTRIBUTION
The goods hold stage is where it is necessary to ensure that the goods remain in good condition, and do not become harmed or damaged through incorrect or unsuitable storage conditions or bad handling. All goods must be stored in a clean, neat, orderly way, in conditions that will not affect their quality or cause them to deteriorate in any way [18].
Goods out might well be the last chance of checking and ensuring that everything is in order before the goods leave a manufacturer’s hands, to the next step in the distribution chain, on their way to the consumer.
It is necessary to make, and keep, a record of each order that is dispatched, which shows:
- Date of dispatch of goods
- Customer’s name and address
- Quantity, name, batch number, and expiry date of each product dispatched
The manufacturer must maintain records of all distribution transactions involving in process or finished goods. All distribution records should be maintained for a minimum 3-year period after the distribution process for any control number has been completed. If expiration dating is used for a product, distribution records must be maintained at least for one year past the expiration date of the product.
CONCLUSION
GMP is a production and testing practice that helps to ensure in built quality product. Many countries have legislated that pharmaceutical companies must follow GMP procedures, and have created their own GMP guidelines that correspond with their legislation. Basic concepts of all of these guidelines remain more or less similar to the ultimate goals of safeguarding the health of the patient as well as producing good quality medicines.
Quality objective can be achieved only through careful planning and implementation of QA system and practical implementation of GMP. The effective implementation of GMP requires extensive care and knowledge about the different components of GMP that should be incorporated form the inception of the manufacturing building and product development till the production.
REFERENCES
1. Lund, W.: Good manufacturing practices. The pharmaceutical codex: principle and practice of pharmaceutics. London, Edition 12, The Pharmaceutical Press, 1994, 362-397.
2. Nally J. and Kieffer R.G.: GMP compliance, productivity and quality, Interpharm, 1998, 465-466.
3. Nally J.D.: Good manufacturing practices for pharmaceuticals, Informa healthcare USA, Inc., ISBN 10:0-8593-3972-3 & ISBN13:, Edition 6, New York, 2007, 978-0-8493-3972- 1.
4. Schedule M. good manufacturing practices and requirements of premises, plant and equipment for pharmaceutical products, 25.03.2012, cdsco.nic.in/html/GMP/ScheduleM(GMP).pdf
5. Asean: Asean operational manual for implementation of GMP, Indonesian national GMP team Asean, 2000.
6. Chaloner-Larsson, G. Anderson, R. Filho, M.A.F.C. & Herrera, J.F.G.: Validation In: A WHO guide to good manufacturing practice (GMP) requirements, World Health Organization, Geneva, 1997.
7. Wikipedia: Good manufacturing practice from Wikipedia, the free encyclopedia, 25.03.2012, en.wikipedia.org/wiki/Good manufacturing practice, 2012.
8. WHOTRS: WHO expert committee on specifications for pharmaceutical preparations: thirty-second report. WHO technical report series: 823, ISBN 92 4140823 6, ISSN Geneva, 1992, 0512- 3054.
9. Eudralex: 2012, Eudralex-Volume 4 Good manufacturing practice (GMP) Guidelines, 23.03.2012, ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm.
10. WHOTRS: Annex 3 WHO good manufacturing practices: main principles for pharmaceutical products, In; WHO expert committee on specifications for pharmaceutical preparations Forty-fifth report WHO Technical report series 961, PP. 94-147, ISBN 978 92 4 120961 8, ISSN, Geneva, 2011, 0512-3054.
11. Sharp J.: Wider aspects of GMP. Good manufacturing practice: philosophy and applications. Illinois, Interpharm Press, 1991, 71-79.
12. US FDA.: Guidance for industry, Quality systems approach to pharmaceutical current good manufacturing practice regulations, Food and drug administration, center for drug evaluation and research (CDER), Center for biologics evaluation and research (CBER), Center for veterinary medicine, office of regulatory affairs (ORA), Rockville, MD, 2004, fda.gov/cvm/guidance/published.html.
13. WHOTR: Annex 2 Supplementary guidelines on good manufacturing practices for heating, ventilation and air-conditioning systems for non-sterile pharmaceutical dosage forms, In; WHO expert committee on specifications for pharmaceutical preparations Fortieth report WHO technical report series 937, Geneva, 2006, 45-84.
14. WHOTR: Annex 3 WHO Good manufacturing practices: water for pharmaceutical use, In; WHO expert committee on specifications for pharmaceutical preparations Thirty-ninth report WHO technical report series 929, 40-58, ISBN 92 4 120929 1, ISSN Geneva, 2005, 0512-3054.
15. Sharp, J.: Quality in the manufacture of medicines and other healthcare products, pharmaceutical press, ISBN London, 2000, 0-85369-431-1.
16. Signore, A. A. and Jacobs, T.: Good design practices for GMP pharmaceutical facilities, Taylor & Francis group, ISBN Boca Raton, 2005, 10: 0-8247-5463-8 & ISBN 13: 978-0-247- 5463-1.
17. 21CFR 211: 2011, PART 211; current good manufacturing practice for finished pharmaceuticals, title 21-food and drugs, chapter I-food and drug administration, Department of health and human services subchapter C-Drugs: General, [Code of federal regulations, title 21, Volume 4, Revised as of April 1, 2011], 21.03.2012,
accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRart=211& showFR=1.
18. Patel K.T. and Chotai N.P.: Documentation and records: Harmonized GMP requirements, Journal of young pharmacists 2011, 3:138-150.
19. Immel, B. K.: 2005, A brief history of the GMPs, Regulatory compliance newsletter, The MP labeling system, (25.03.2012).
20. Sharp, J.: Good Pharmaceutical manufacturing practice rationale and compliance, CRC Press, ISBN Washington, D.C., 2005, 0-8493-1994-3.
21. US FDA: Improving public health through human drugs, CDER 2001 Report to the nation, Food and drug administration, Center for drug evaluation and research, 2001.
22. Wikipedia: Three-phase electric power from Wikipedia, the free encyclopedia, 01.05.2012, en.wikipedia.org/wiki/Three phase electric power, 2012.
23. PIC/S Secretariat (Ed.): guide to good manufacturing practice for medicinal products, Pharmaceutical inspection convention/ pharmaceutical inspection co-operation scheme PE 009-2 1 July 2004, Geneva.
24. Huber, L. (2012). Validation of analytical methods and procedures, 25.03.2012, labcompliance.com/tutorial/methods/default.aspx.
REFERENCE ID: PHARMATUTOR-ART-2245
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 9 Received On: 06/07/2014; Accepted On: 15/07/2014; Published On: 01/09/2014How to cite this article: VK Chaudhari, V Yadav, PK Verma, AK Singh; A Review on Good Manufacturing Practice (GMP) for Medicinal Products; PharmaTutor; 2014; 2(9); 8-19 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








