
ABOUT AUTHOR:
Mr. Vivek P. Chavda, Dr. Moinuddin M. Soniwala
Department of Pharmaceutics, B.K. Mody Government Pharmacy College,
Rajkot – 360003, Gujarat (India)
vivek7chavda@gmail.com
INTRODUCTION1,2
In recent years, many important initiatives have been undertaken by regulatory authorities and industry associations to promote international harmonization of regulatory requirements. International Conference on the Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) was organized in April 1990 and has as its sole and primary purpose the creation of international standards for the purpose of pharmaceutical research. This process was initiated in order to harmonize the submission requirements for new pharmaceuticals in the three main regions of Europe, the United States, and Japan and to avoid duplication, inefficiencies and delays.
The six cosponsors of ICH were
- European Commission,
- European Federation of Pharmaceutical Industry Association (EFPIA),
- Japanese Ministry of Health (MHW),
- Japanese Pharmaceutical Manufacturers Association (JPMA),
- Food and Drug Association (FDA), and the Pharmaceutical Research
- Manufacturers of America (PhRMA)
Reference ID: PHARMATUTOR-ART-1950
The ICH Steering Committee includes representatives from each of the ICH sponsors and the IFPMA, as well as observers from the World Health Organization, the Canadian Health Protection Branch, and the European Free Trade Area. In the Federal Register of March 7, 1996 (61 FR 9310), FDA published a draft tripartite guideline entitled ‘‘Guideline for the Photostability Testing of New Drug Substances and Products.’’ The notice gave interested persons an opportunity to submit comments by June 5, 1996.
After consideration of the comments received and revisions to the guideline, a final draft of the guideline was submitted to the ICH Steering Committee and endorsed by the three participating regulatory agencies at the ICH meeting held on November 5, 1996. In the Federal Register of September 22, 1994 (59 FR 48754), the agency published a guideline entitled ‘‘Stability Testing of New Drug Substances and Products.’’ The guideline addresses the generation of stability information for submission to FDA in new drug applications for new molecular entities and associated drug products. In the discussion of ‘‘stress testing’’ for both drug substances and drug products, the guideline states that ‘‘light testing’’ should be an integral part of stress testing and will be considered in a separate ICH document.
HISTORY2,3
Prior to 1960s there were not many controls over introduction of new drugs and also over the assurance of the quality by the manufacturer over his established drug products. Around 1970s the pharmaceutical industry started getting global but the registration of medicines remained a national responsibility.Although the laws of all the countries were based on the same fundamental obligations to evaluate the quality, safety and efficacy the detailed technical requirements differed from country to country. So the companies had to duplicate many time consuming and expensive test procedures, in order to market new products, internationally. All this resulted in unnecessary expenses and long delays in introducing new drugs. Hence a necessity to harmonize or make uniform, the testing procedures and regulatory requirements of different countries was felt and the result is the birth of ICH in April 1990.
AIM2,3
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) is a unique project that brings together the regulatory authorities of Europe, Japan and the United States and experts from the pharmaceutical industry in the three regions to discuss scientific and technical aspects of product registration.
Objectives of ICH
- More economical use of human, animal, and material resources.
- Elimination of unnecessary delay in the global development & availability of new medicines.
- Maintaining safeguards on Quality, safety & efficacy, and regulatory obligations to protect public health.
The ICH topics are divided into four categories and ICH topic codes are assigned according to these categories
1. QUALITY GUIDELINE
Those relating to chemical and pharmaceutical Quality Assurance (Stability Testing, Impurity Testing, etc.)
2. SAFETY GUIDELINE
Those relating to in vitro and in vivo pre-clinical studies (Carcinogenicity Testing, Genotoxicity Testing, etc.)
3. EFFICACY GUIDELINE
Those relating to clinical studies in human subject (Dose Response Studies, Good Clinical Practices, etc.)
4. MULTIDICIPLINARY GUIDLINE
Cross-cutting Topics which do not fit uniquely into one of the above categories (MedDRA, ESTRI, M3, CTD, M5)
1) QUALITY:
Q1A (R2): Stability testing of new drug substances and products.(Revised guideline)
Q1B:Photostability testing.
Q1C: Stability testing of new dosage forms.
Q1D: Bracketing & Matrixing designs for stability testing of new drugs substances and products.
Q1E: Evaluation of Stability data.
Q1F: Stability Data Package for Registration Applications in Climatic Zones III and IV
Q1B:2
“PHOTOSTABILITY TESTING OF NEW DRUG SUBSTANCES AND DRUG PRODUCTS"
CONTENT
1. GENERAL
A. PREAMBLE
B. LIGHT SOURCES
C. PROCEDURE (DECISION FLOW CHART)
2. DRUG SUBSTANCES
A. PRESENTATION OF SAMPLES
B. ANALYSIS OF SAMPLES
C. JUDGEMENT OF RESULTS
3. DRUG PRODUCTS
A. PRESENTATION OF SAMPLES
B. ANALYSIS OF SAMPLES
C. JUDGEMENT OF RESULTS
4. ANNEX
A. QUININE CHEMICAL ACTINOMETRY
1. GENERAL
This document is an annex to the ICH parent stability guideline and addresses the recommendations on what should be submitted regarding stability of new dosage forms. The light testing is an integral part of the stress testing.
NEW DOSAGE FORMS
Stability protocols for new dosage forms should follow the guidance in the parent stability guideline. However, a reduced stability database at submission time (e.g., 6 months accelerated and 6 months long term data from ongoing studies) may be acceptable in certain justified cases.
A. PREAMBLE
The intrinsic photostability characteristics of new drug substances and products should be evaluated to demonstrate that, as appropriate, light exposure does not result in unacceptable change.The guideline does not cover the photostability of drugs after administration.Normally, photostability testing is carried out on a single batch of material selected as described under Selection of Batches in the Parent Guideline. Under some circumstances these studies should be repeated if certain variations and changes are made to the product (e.g., formulation, packaging).
Example4: Nifedipine (NIF) a 1, 4-dihydropyridine calcium channel antagonist, undergoes photodegradation to nitroso analogues of dihydronifedipine (NDNIF) when exposed to sunlight. Photodegradation products of NIF have no clinical activity, so different formulations of NIF must remain unchanged. If NIF preparations become unstable in exposure to light, they could cause therapeutic failure. The present study was carried out in order to investigate the photostability of commercially available NIF products.
A systematic approach to photostability testing is recommended covering, as appropriate, studies such as:
[1] Tests on the drug substance;
[2] Tests on the exposed drug product outside of the immediate pack; and if necessary;
[3] Tests on the drug product in the immediate pack; and if necessary ;
[4] Tests on the drug product in the marketing pack.
WAYS FOR STABILIZATION5: Suitable packing Photo stabilizer (Light absorber) Protection of drug from light during mfg. Coating. Eg. Photo stabilization of Molsidomine Tablet;Molsidomine Morpholine dvt. (Potential carcinogenic) It was stabilized by;
* Incorporation of light absorbing excipients. colorants – curcumine and azorubine
* Incorporation of pigments. (eg. TiO2 and ZnO3)
* By coating a) white coating ( 4.8% TiO2)
b) colored coating ( yellow & red iron oxide added to std. coating containing 4.8%TiO2)
Effect of excipient on photostability
Normally absorbing excipient in film coating has photoprotective action but in some cases negative effect has been reported.Eg. The formation of peroxide, influence through buffer substances and discoloration of aromatic ingredient
B. LIGHT SOURCES
The light sources described below may be used for photostability testing. The applicant should either maintain an appropriate control of temperature to minimize the effect of localized temperature changes or include a dark control in the same environment unless otherwise justified.For both options 1 and 2, a pharmaceutical manufacturer/applicant may rely on the spectral distribution specification of the light source manufacturer.
Option 1
Any light source that is designed to produce an output similar to the D65/ID65 emission standard such as an artificial daylight fluorescent lamp combining visible and ultraviolet (UV) outputs, xenon, or metal halide lamp. D65 is the internationally recognized standard for outdoor daylight as defined in ISO 10977 (1993). ID65 is the equivalent indoor indirect daylight standard.For a Light source emitting significant radiation below 320 nm, an appropriate filter(s) may be fitted to eliminate such radiation.
Option 2
For option 2 the same sample should be exposed to both the cool white fluorescent and near ultraviolet lamp.
[1] A cool white fluorescent lamp designed to produce an output similar to that specified in ISO 10977(1993) ; and
[2] A near UV fluorescent lamp having a spectral distribution from 320 nm to 400 nm with a maximum energy emission between 350 nm and 370 nm; a significant proportion of UV should be in both bands of 320 to 360 nm and 360 to 400 nm.
C. PROCEDURE
For confirmatory studies, samples should be exposed to light providing an overall illumination of not less than 1.2 million lux hours and an integrated near ultraviolet energy of not less than 200 watt hours/square meter to allow direct comparisons to be made between the drug substance and drug product.Samples may be exposed side-by-side with a validated chemical actinometric system to ensure the specified light exposure is obtained, or for the appropriate duration of time when conditions have been monitored using calibrated radiometers/lux meters.If protected samples (e.g., wrapped in aluminum foil) are used as dark controls to evaluate the contribution of thermally induced change to the total observed change, these should be placed alongside the authentic sample.
Decision flow chart for Photostability testing of drug products
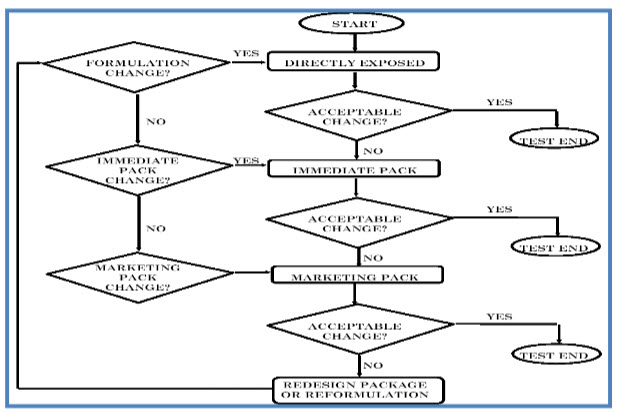
2. DRUG SUBSTANCES
For drug substances, Photostability testing should consist of two parts:
1]Forced degradation testing
2] Confirmatory testing.
The purpose of forced degradation testing studies is to evaluate the overall photosensitivity of the material for method development purposes and/or degradationpathway elucidation. This testing may involve the drug substance alone and/or in simple solutions/ suspensions to validate the analytical procedures.In these studies, the samples should be in chemically inert and transparent containers. In these forced degradation studies, a variety of exposure conditions may be used, depending on the photosensitivity of the drug substance involved and the intensity of the light sources used.Under forcing conditions, decomposition products may be observed that are unlikely to be formed under the conditions used for confirmatory studies. Confirmatory studies should then be undertaken to provide the information necessary for handling, packaging, and labeling (see section I.C., Procedure, and II.A. Presentation for information on the design of these studies).
A. Presentation of Samples
Care should be taken to ensure that the physical characteristics of the samples under test are taken into account and efforts should be made, such as cooling and/or placing the samples in sealed containers, to ensure that the effects of the changes in physical states such as sublimation, evaporation or melting are minimized.All such precautions should be chosen to provide minimal interference with the exposure of samples under test. Possible interactions between the samples and any material used for containers or for general protection of the sample should also be considered and eliminated wherever not relevant to the test being carried out.As a direct challenge for samples of solid drug substances, an appropriate amount of sample should be taken and placed in a suitable glass or plastic dish and protected with a suitable transparent cover if considered necessary. Solid drug substances should be spread across the container to give a thickness of typicallynot more than 3 mm. Drug substances that are liquids should be exposed in chemically inert and transparent containers.
B. Analysis of Samples
At the end of the exposure period, the samples should be examined for any changes in physical properties (e.g., appearance, clarity, or color of solution) and for assay and degradants by a method suitably validated for products likely to arise from photochemical degradation processes.Where solid drug substance samples are involved, sampling should ensure that a representative portion is used in individual tests. Similar sampling considerations, such as homogenization of the entire sample, apply to other materials that may not be homogeneous after exposure. The analysis of the exposed sample should be performed concomitantly with that of any protected samples used as dark controls if these are used in the test.
C. Judgment of Results
The forced degradation studies should be designed to provide suitable information to develop and validate test methods for the confirmatory studies. These test methods should be capable of resolving and detecting photolytic degradants that appear during the confirmatory studies. When evaluating the results of these studies, it is important to recognize that they form part of the stress testing and are not therefore designed to establish qualitative or quantitative limits for change.The confirmatory studies should identify precautionary measures needed in manufacturing or in formulation of the drug product, and if light resistant packaging is needed. When evaluating the results of confirmatory studies to determine whether change due to exposure to light is acceptable, it is important to consider the results from other formal stability studies in order to assure that the drug will be within justified limits at time of use (see the relevant ICH Stability and Impurity Guidelines).
3. DRUG PRODUCT
(It is same as that described in drug substances)
4. ANNEX
A. Quinine Chemical Actinometry6
The following provides details of an actinometric procedure for monitoring exposure to a near UV fluorescent lamp (based on FDA/National Institute of Standards and Technology study). For other light sources/actinometric systems, the same approach may be used, but each actinometric system should be calibrated for the light source used.Prepare a sufficient quantity of a 2 per cent weight/volume aqueous solution of quinine monohydrochloride dihydrate (if necessary, dissolve by heating).
Option 1: Use 20 ml colourless ampoules (seal hermetically).
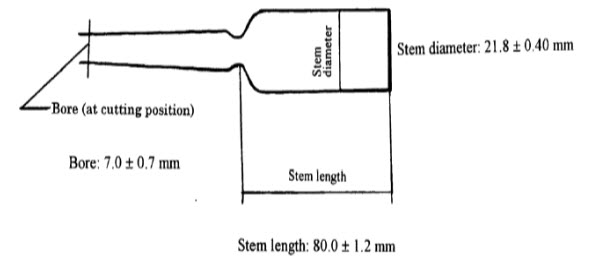
Figure 1: Shape and Dimensions for ampoule specifications
Option 2: Use 1 cm quartz cell.
For both the options, prepare sample and control wrap in aluminum foil to protect completely from light, and measure their absorbance At and Ao respectively at 400nm using 1cm path length. Measure the change in absorbance.The length of exposure should be sufficient to ensure a change in absorbance of at least 0.9.
REFERENCE
1. Swarbrick J, Photo stability, Encyclopedia of Pharmaceutical Technology, Volume 19: 227-235.
2. Department of health andhuman services,Federal Register, Vol. 62, No. 95 / Friday, May 16, 1997 / Notices (Accessed on 7th July, 2013)
3. ICH Harmonised Tripartitie Guidelines, Guideline for the Photostability Testing of New Drug Substances and Products; Availability(Q1B); ich.org, 2013: 1-12.
4. Katayoun J,Ramin M,Photostability Determination of Commercially Available Nifedipine Oral Dosage Forms, Iraniann Journal of Pharmaceutical Research.2003, 2(2) :111-115.
5. Drug Stability: Principles and Practices, 3rd Edition, edited by Jens T. Carstensen and C.T. Rhodes; Chapter-13 & 17.
6. Yoshioka S. Quinine Actinometry as a method for calibrating ultraviolet radiation intensity in light-stability testing of pharmaceuticals. Drug Development and Industrial Pharmacy,1994, 20 (13): 2049 – 2062.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








