
 ABOUT AUTHORS:
ABOUT AUTHORS:
DEVENDRA SINGH1*, PANKAJ KUMAR SHARMA1, ROOHI KESHARWANI2, Dr. UDAI VIR SINGH SARA1
1Raj kumar goel institute of technology, Delhi-Meerut Road, Ghaziabad, India
2Chandra shekhar singh college of pharmacy, Kaushambi, Uttar Pradesh, India.
* devendrasingh.pisces@gmail.com
ABSTRACT:
Oral administration still dominates drug therapy and more than 60 % of marketed drugs are oral products. This type of drug administration is preferred due to its convenience, high patient compliance, less stringent production conditions and lower costs. Unfortunately, this traditional drug delivery method has its limitations, due to gastrointestinal permeability, metabolism and elimination of drugs by the liver or gastrointestinal mucosa (first-pass effect). The main drawback of the oral route is that only those compounds that are stable in the gastrointestinal tract can be administered in this way. For this reason, the oral route has been used for mainly non-peptide drugs. Delivery of a drug by oral route is predominantly restricted by pre-systemic degradation and poor penetration across the gut wall. The major challenge in the oral drug delivery is the development of novel dosage forms to endorse absorption of poorly permeable drugs across the intestinal epithelium. In this article we reviewed the various permeation enhancers, histology of small intestine, barriers, application, advantages, some basic permeability enhancement techniques which are useful for enhancing the permeability of poorly permeable drugs
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1680
INTRODUCTION
The traditional drug administration routes used were oral administration for systemic effects and topical for local effects. Drugs could also be self-administered by inhalation, suppository and sometimes injections. The other routes of delivery usually required the intervention of a healthcare provider and the pain, fear and the possibility of infections associated with injections often resulted in low patient compliance and therefore have aided the development of suitable non-parenteral routes of administration. Oral administration still dominates drug therapy and more than 60 % of marketed drugs are oral products . This type of drug administration is preferred due to its convenience, high patient compliance, less stringent production conditions and lower costs. Delivering a drug by oral route is also preferred for its convenience. Tablets and capsules can be prepared in large quantity at low price. Therefore in lead optimization step of drug discovery, oral bioavailability of a drug is important. It depends on various factors the most common being intestinal permeability, solubility during gastrointestinal transit, liberation from dosage form, liability to efflux and metabolism. Development in the field of combinatorial chemistry and high throughput screening has made it possible to generate a large number of drug candidates but it has also resulted in a number of poorly soluble and or poorly absorbable drugs. A new trend of drug development based on pharmacogenomics or development of molecular targeted drugs is also encouraging the tendency, and it does not necessarily lead to good output in terms of new drug development. Therefore it is necessary to improve the membrane permeability as well. The pharmacokinetic profile of a drug is dependent on the drugs ability to crossbiological membranes. The permeability characteristics of compounds therefore affect its absorption, distribution and elimination (Mälkiä et al., 2004). It is estimated that approximately 40 % of the failures in drug development programs during clinical phases are due to problems in pharmacokinetics and drug delivery (Kennedy, 1997). The absorption of an orally administered compound depends on many different parameters, such as the chemical structure of the drug, permeability of the intestine brush border membrane (BBM), intestinal motility, gastrointestinal transit times, fluid volume, bile salt composition, enzyme systems, food and the physiological state of the human intestinal tissue. It is therefore clear that the absorption of a drug by the intestine is a complex process (Oulianova et al., 2007; Martinez et al., 2002). All drugs are now classified according to the biopharmaceutical classification BCS into four categories on the basis of solubility and permeability to rationalize science of drug delivery and simplify complications in the drug registration of newly evolving diverse compounds for regulatory authorities. Among the different classes of BCS the per oral delivery of class 3 and 4 drugs is partially or completely decreased due to their poor intestinal permeability. A great number of currently available drugs fall under the class III of the biopharmaceutical classification system, possess high therapeutic potential but cannot be delivered by oral route because of its poor permeation across the GIT epithelia. It is important that the limiting factor to intestinal absorption can be modelled when using in vitro models to predict in vivo performance. Release of the active compound and absorption must occur within the available transit time. The drug must be released before reaching the absorptive site of the GIT and must be stable in the luminal fluids (Dressman and Reppas, 2000). The physicochemical properties of an orally administered drug are generally the major determinants of intestinal permeability. This includes molecular size and shape, pKa, lipophilicity (log P/log D), charge/ionisation and hydrogen bonding properties. Solubility of the compound in the intestinal tract is an extremely important factor that dictates the dissolution characteristics of the drug and eventually influences the bioavailability of the compound. Even though the underlying driver for solubility in the gastrointestinal fluids is the aqueous solubility of the drug, the solubility of the drug in the GIT may be influenced by pH profile, solubilisation via naturally occurring surfactants and food components, as well as complex formation with food and native components of the gastrointestinal contents (Dressman et al., 2007).
Biopharmaceutical Classification System
The BCS is a scientific framework for classifying a drug substance based on its aqueous solubility and intestinal permeability .When combined with the in vitro dissolution characteristics of the drug product, the BCS takes into account three major factors: solubility, intestinal permeability, and dissolution rate, all of which govern the rate and extent of oral drug absorption from IR solid oral-dosage forms (Amidon GL et al, 1995, 12, 413–420.)
(Table 1): According to the BCS the drugs can be categorized in to four basic groups on the bases of their solubility and permeability GIT mucosa.
|
Class |
Permeability/ Solubility |
Absorption rate control step |
|
Class I |
High /High |
Gastric emptying |
|
Class II |
High /Low |
Dissolution |
|
Class III |
Low /High |
Permeability |
|
Class IV |
Low /Low |
Case by case |
For Class III drugs permeability is rate limiting step for drug absorption. These drugs exhibit a high variation in the rate and extent of drug absorption. Since the dissolution is rapid, the variation is attributable to alteration of physiology and membrane permeability rather than the dosage form factors. These drugs are problematic for controlled release development. These drugs showed the low bioavailability and need enhancement in permeability. Following permeation enhancers can be used .(P. Anilkumar et.al,2000)
TABLE.2. Classification of intestinal permeation enhancers:
|
Surfactants |
Ionic: Sodium lauryl sulphate Sodium dodecylsulphate Dioctyl sodium sulfosuccinate Nonionic: Polysorbitate Nonylphenoxypolyoxetyylenes Tween80 |
|
Bilesalts & its derivative |
Sodium glycholate Sodium deoxycholate Sodium taurocholate Sodium dihydrofusidate Sodium glycodihdro fusidate Sodium glycolate |
|
Fattyacids & its derivatives |
Oleic acid Caprylic acid Lauric acids Sodium caprate Acyl carnites Acyl choline Sodium caprylate |
|
Chelating agents |
EDTA Citric acid Salicylates |
|
Chitosans & derivatives |
N-sulfanto-N,O-carboxymethylchitosan N-trimethylated chloride(TMC) Chitosan glutamate |
|
Other enhancers |
Zonula occludens toxin (Zot) polycarbophyl-cysteine conjugate(PCP-Cys) |
DETERMINING DRUG SUBSTANCE PERMEABILITY CLASS:
The following methods can be used to determine the permeability of a drug substance from the gastrointestinal tract:
· In vivo intestinal perfusions studies in humans.
· In vivo or in situ intestinal perfusion studies in animals.
· In vitro permeation experiments with excised human or animal intestinal tissue.
· In vitro permeation experiments across epithelial cell monolayer
[adsense:468x15:2204050025]
HISTOLOGY: SMALL INTESTINE:
The walls of the small intestine are composed of four layers of tissue: mucosa, submucosa, muscularis externa and the serosa (Fig. 42). The mucosa is composed of the intestinal epithelium, lamina propria and muscularis mucosae. The mucosa has a characteristic villous form in the whole of the small intestine, with short glands known as crypts of Lieberkühn between the villi, extending down to the muscularis mucosae. The villi are finger-like projections of the lamina propria that is the connective tissue layer of the mucosa. The villi tend to be longest in the duodenum and become shorter towards the ileum. The mucosa and submucosa are thrown up into circularly arranged folds, called plicae circulars or valves of Kerkring, which are particularly numerous in the jejunum. Plicae circulares are only found in the jejunum and ileum The epithelium of the mucosa consists of simple columnar epithelium or enterocytes,mucous/goblet cells, Paneth cells, stem cells and endocrine cells. The enterocytes are responsible for digestion and absorption and cover the villi and crypts. Themucous/goblet cells, Paneth cells, stem cells and endocrine cells are dispersed betweenthe enterocytes and their numbers and distribution vary in the different zones of thesmall intestine. The proportion of goblet cells in the epithelium increases distally.Numerous microvilli are present at the luminal surface of the enterocytes and since themicrovilli project from the epitheliums free border like the bristles on a brush, theseepithelial cells are said to have a brush border. The connection tissue of the laminapropria contains an arteriole, a venule, a capillary network and a lacteal, which is alymphatic vessel (Fig. 43). Nutrients and drugs pass through the epithelial cells coveringthe villus and then pass through the capillary walls and the lacteal to enter the blood andlymphatic fluid. The entire epithelium of the intestine is replaced every 3 to 5 days.Another prominent feature of the small intestine is lymphoid aggregations known as Peyer's patches within the lamina propria. These structures are associated with thefunction of the immune system (Martini, 2006; Tortora and Grabowski, 1993). Themuscularis mucosa, a layer of circular smooth muscle fibres, lies immediately beneaththe mucosal crypts and separates the mucosa (lamina propria) from the submucosa.The submucosa is vascularised loose connective tissue containing larger blood vessels,lymphatic vessels and adipose cells as well as submucosal (Meissner's) plexuses. Itextends into and forms the core of the plicae circulares. Beneath the mass ofsubmucosal glands is the muscularis externa, which is composed of an inner circularlayer and an outer longitudinal layer of smooth muscle. These two muscle types areresponsible for peristaltic activity. Between the two layers of smooth muscle is themyenteric plexus (plexus of Auerbach). The peritoneal aspect of the muscularis externais invested by the loose collagenous serosal, which is lined on its peritoneal surface bymesothelium, that is identical in appearance to the mesothelium lining of the pleura.
If the small intestine was a simple tube with smooth walls it would have had a total areaof roughly 3300 cm2, but the surface area for digestion and absorption is increased to afactor of more than 600 (2 million cm2) due to the plicae and the forest of villi andmicrovilli (Martini, 2006; Tortora and Grabowski, 1993).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
PHYSIOLOGY OF BARRIERS:
· Tight junctions and epithelial barrier function:
Tight junctions restrict epithelial cells immediately below the brush border forming a seal between neighboring epithelial cells. This seal acts as a gate to restrict passage of small molecules in a charge specific manner and completely occludes diffusion of molecules with molecular radii larger than 0.1nm. In addition, the tight junction acts as a fence that separates components of the apical and basolateral domains of the epithelial plasma membrane joined at their apical surfaces by tight junctions BY[11](Itoh H et al,96:1721–1726.).
· Biochemical composition of the tight junctions:
The biochemical composition of tight junctions is still being elucidated, but many of the key components have been identified. It is currently recognized that the tight junctions primarily are complex multicomponent protein structures. Identification of the principal transmembrane component has only recently come to light and it is now known that the tight junction is composed of a homotypic protein termed occludin. Other tight junctional complex proteins which have been identified are ZO-15, claudin. All of these proteins are oriented peripheral to the cytoplasmic surface of the tight junction complex and are thought to be involved in the stabilization and/or regulation of tight junction integrity.BY (Soboll S et al,2000)
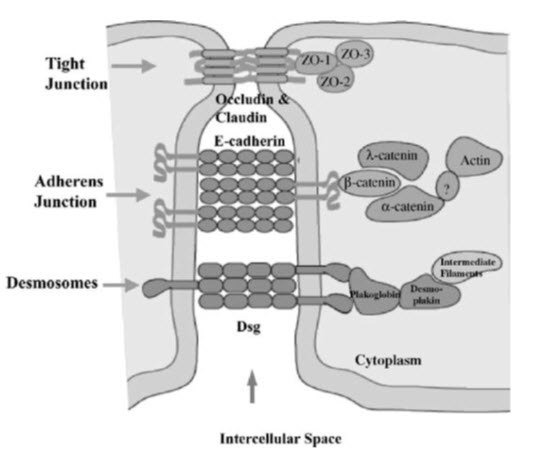
Figure 1: Biochemical composition of tight junction
Regulation of tight junction permeability:
The tight junction complex is not a static structural component as once was thought, but slightly resembles a dynamic and elaborate protein signaling complex. Regulation of tight junction paracellular permeability by various physiological, pathological, and experimental agents has been extensively examined in a number of in situ and in vitro culture models, particularly Caco-26, brain endothelial, and MDCK cells. Peptide hormones, cytoskeleton perturbing agents, oxidants, Ca++ chelators and ionophores has all been shown to alter paracellular permeability by disrupting tight junctions. In addition, it has been determined that tight junction permeability is influenced by nearly all second messenger and signaling pathways, such as tyrosine kinases, Ca++, protein kinase C (PKC), protein kinase A (PKA), G proteins, calmodulin, CAMP, and phospholipase C.. Two factors which appear to play a prominent role in the regulation of paracellular permeability by absorption enhancers are contraction of the peri junctional actin myosin ring, and protein kinase or phosphatase mediated changes in tight junction protein phosphorylation.
Role of the perijunctional actin myosin ring:
Adjacent to the tightjunction in the cytoplasm is an actinmyosinring which restricts the cell. Thisring is associated with both the tight andintermediate junctional complexes and cancontract exerting an inward force on thelateral plasma membrane. Suchcontractions are ATP-dependent and havebeen correlated with a loosening of thetight junctions indicating that contractionsof the perijunctional ring pull on tightjunction components and induce changesin paracellular permeability betweenneighboring cells. Further evidence for aphysical link between the perijunctionalactin-myosin ring and tight junctions hasbeen inferred by direct observation of tightjunction-associated actin and byobservations showing disruption in thestructure and integrity of tight junctions byagents which disrupt actin filaments (e.g.cytochalasin D) possible to visualize theperijunctional actin-myosin ring by staining actin filaments with fluorescentlabeled phalloidin7. Using this approach global changes in actin distribution have been documented with some tight junction disrupting agents including oxidants, protein kinase C activators , Ca++ depletion , and cytoskeleton disrupting agents while more subtle changes have been observed with other tight junction disrupting agents (i.e. interferon-y).
Role of calcium:
Extracellular calcium levels play a prominent role in the formation and regulation of tight junctions and paracellular permeability. Adhesion at the adherens junction is mediated by cadherins7 which are Ca’ ’ -dependent,cell-cell adhesion molecules that interact Homo typically. Removal of Ca’ + has been known for many years to lead to an increase in tight-junction permeability and cause a redistributionof tight junction proteins. It appears that it is the disruption of cadherin adhesiveness by removal of Cat+ rather than a direct effect on the tight junction, which leads to the increase in paracellular permeability.(P. Anilkumar et al ,2000)
Role of CAMP:
Intracellular CAMP8 alters paracellular permeability by reducing NaCl diffusion potentials and increase passive permeability to Cl- as well as Cl: Na permeability ratios in intestinal and gall bladder epithelium. CAMP may also decrease tight junction resistance, but this effect may be masked by the increased resistance that accompanies collapse of the lateral spaces.
Role of ATP depletion:
Under normal physiological conditions, the tight junction is maintained by an energy-dependent (ATP) process involving the actin cytoskeleton and tight junctions. Alteration in cellular energy status, a decrease in adenosinetriphosphate (ATP) levels, has been shown to disrupt epithelial barrier function andincrease permeability. Energy depletion results in net loss of phosphorylation of brush border, and possibly junctional, proteins.
DRUG ABSORPTION FROM THE GASTRO-INTESTINAL TRACT:

Fig 2: Drug absorption from the GIT.
Mechanisms of intestinal drug permeability:
The intestinal mucosa ca be considered as a system of sequential barriers to drug absorption, the outermost barrier being the mucus layer and the unstirred water layer. The gel structure of the mucus is thought to be a barrier to absorption of highly lipophilic drugs and some peptides because of the restricted diffusion in this matrix. The absorptive epithelium lining the GI tract follows the folds and villi that increase the anatomical surface area of the mucosa several-fold in the small intestine. The villi are interspaced with crypts in which the regeneration of intestinal cells occurs. In between the crypts and the tips of the villi are the basal parts of the villi. The properties which are relevant for drug absorption differ between the cells along the crypt-villus axis BY[13](Artursson P.et al ; 1990 )
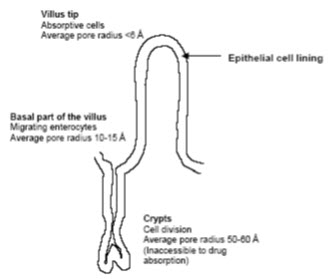
Figure- 3: The absorptive epithelium lining of the GI tract
Schematic illustration of the different transport routes that are relevant for drug absorption. 1. Passive transcellular and 2. Parcellular transport, 3. Carrier-mediated efflux and 4. Carrier mediated active transport.The intestinal epithelium is a gatekeeper, i.e. it controls the entry of nutrients and xenobiotics (for example, medicines). Knowledge of the absorption and metabolism of these substances at the intestinal mucosal level is of particular importance, since the oral bioavailability of a drug is defined as the fraction of an oral dose that reaches the systemic circulation. Drug absorption is considered to be a complex transfer process across the intestinal lining, which includes passive diffusion through the paracellular space and/or membranes of absorptive cells, vesicular uptake (endocytosis/pinocytosis), and release at the basolateral space (transcytosis). This transport may or may not be receptor-mediated (or transporter mediated), entailing uptake across the apical domain with subsequent passive diffusion into the basolateral space (Figure 1; 3). Each transport mechanism depends on the physicochemical properties of the absorbed compound, such as its stereochemistry, partition into membranes, molecular weight and/or size, molecular volume, pKa, solubility, chemical stability and charge distribution. Physiological factors such as gastric emptying, gastrointestinal motility, intestinal pH, blood flow, lymph flow , pathological state, drug interactions, nutrition, and mucus dissolution, also need to be considered when evaluating absorption (Pade, V. & Stavchansky, S. (1998).
Site of absorption
Drugs are absorbed throughout the GIT. Absorption of drugs throughout the GIT varies with location. The stomach is the first site for drug release and absorption. The pH of the stomach in the fasted state usually lies in the range of pH 1-3. After a meal the pH, increases to values up to 7 due to the buffering effects of the food. For drugs that are highly soluble at a gastric pH, complete dissolution can occur in the stomach, but for weak acids little dissolution will occur in the stomach, while high dissolution will occur in the small intestine due to this regions higher pH. Poorly soluble neutral compounds have slow dissolution in the gastric region and often dissolution is not completed before the drug reaches the first absorptive sites in the small intestine. For poorly soluble weak bases, solubility is likely to be higher in the stomach than elsewhere in the GIT. This may result in supersaturation, but precipitation of the drug is hindered by bile components as the drug moves out of the stomach into the higher pH of the small intestine (Dressman et al., 2007). The pH of the small intestine is 7-7.8 (Bajpai et al., 2003). In the fasted state, it may be below neutral (pH 6-6.5) (Dressman et al., 1990). The proximal small intestine's pH is influenced more by food intake than the pH of the distal regions. Generally, the upper part of the small intestine is considered to have a higher capacityfor drug absorption than the lower part. This is due to an increase in tightness of the epithelium in the distal intestinal area, as well as a decrease in total surface area. The luminal pH of the different areas also plays a role. In mammals/humans the jejunum is generally the major absorbing site for most drugs. It has the largest surface area and it is the site of the most active carrier-mediated transport in the gut (Kasim et al., 2004; Sun et al., 2002). The villous structures in the upper intestine enlarge the surface area of the intestine membrane. Villous structures of the jejunum amplify the area four-fold compared to the colon and two-fold compared to the ileum. It seems that this area is relatively leaky and therefore contributes to the high permeability of hydrophobic drugs in the upper intestinal membrane. However, some drugs have been reported to exhibit high permeability even to the lower part of the intestine (ileum and colon) (Balimane et al., 2000). Generally, the permeability of the colonic membrane is much lower than that of the small intestine, but some drugs are known to be absorbed from the colon.
Absorption and solubility:
The epithelial cells lining the gastrointestinal tract are the major barrier to absorption of many orally administered drugs (Jackson, 2005). They are a heterogeneous population of cells that include enterocytes/absorptive cells, goblet cells that secrete mucin, endocrine cells, Paneth cells, M cells, tuft cells and cup cells (Madara and Trier, 1987; Carr and Toner, 1984). Enterocytes make up 90 % of the cells in the epithelium and are Chapter 4: Small intestine mucosa 132 responsible for the majority of the absorption of both nutrients and drugs in the small intestine. The epithelial membrane controls the transport of both low and high permeability compounds regardless of the transport mechanism in vivo (Lennernäs, 2007). It is highly polarised with distinct apical and basolateral membranes that are separated by tight junctions (Balimane et al., 2000). The basolateral surface faces the bloodstream and the apical surface faces the intestinal lumen. The membranes around the intestinal cells are lipid bilayers containing proteins (e.g. receptors and carrier molecules). The mucus layer at the surface of the intestinal epithelium exhibits a rate-limiting barrier, especially against the absorption of highly permeable drugs. The thickness of the mucus layer varies in different luminal segments (Winne and Verheyen, 1990). The mucous gel layer consists of a three-dimensional network of glycoproteins and the extracellular matrix comprises tight junctions, which function is regulated by different transmembrane and intracellular proteins (Ho et al., 2004). The small intestines primary source of solubilisation is the bile components. The bile salt conjugates, phospholipids and cholesterol combine (additionally with lipolysis products in the fed state) to create mixed micelles that can solubilise lipophilic molecules and correlations have been established for solubilisation by mixed micelles as a function of logP for neutral compounds (Dressman et al., 2007). The rate and extend of drug absorption from a solid dosage form during its transit through the small and large intestine depends on several factors: drug release and dissolution kinetics, stability and binding properties in the lumen, gastrointestinal transit time and intestinal permeability (Lennernäs, 2007). Permeation of compounds across the intestinal membrane is a complex and dynamic process. Two fundamental processes determine oral drug absorption: the drugs solubility or dissolution rate in gastrointestinal fluids and its gastrointestinal permeability through the lipid bilayers of the epithelial cells lining the intestinal wall. However, the permeability of a drug is not a simple two-step process of solubilisation and diffusion, but rather consists of a variety of complex molecular events (Pleuvry, 2005).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Different types of permeation routes:
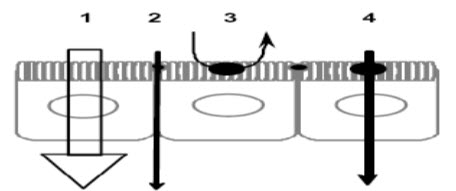
FIGURE 4:1) Paracellular; 2) transcellular passive diffusion; 3) transcytosis; 4) carrier-mediated uptake at the apical domain followed by passive diffusion across the basolateral membrane.
* Passive transcellular transport:
Drug transport via the passive transcellular route requires that the solute permeates the apical cell membrane. Cell membranes are made up of phospholipids arranged in bilayers that are intermingled with membrane proteins. The composition of phospholipids and proteins varies from cell type to cell type and may theoretically give rise to different permeability properties depending on the cell type. In addition, the intestinal enterocytes have a polarized cell membrane with distinct differences in membrane composition in the apical and the basolateral membrane. It is generally believed that the apical membrane has a lower permeability than the basolateral membrane and the former is therefore considered to be the rate limiting barrier to passive transcellular drug transport.
* Active transport:
Transport proteins embedded in the apical cell membrane actively shunt nutrients such as peptides, amino acids and sugars across the phospholipid bilayer. In order to restrict access of unwanted solutes via this pathway, these transporters display substrate specificity. Therefore, in order to utilize this pathway to increase absorption, the drug has to share some structure similarity with the normal substrate. A limited number of drugs are substrates for uptake carrier proteins. These include some cephalosporin antibiotics, cytostatics and angiotensin-converting enzyme (ACE) inhibitors that are substrates for oligopeptide transporters. The oligopeptide transporters have unusually broad substrate specificity, are abundantly expressed in the small intestine and have toxic substances and also, to eliminate such substances from the blood.
* Paracellular transport:
Drugs of small to moderate molecular weights (MWs) can permeate the intestinal epithelium through the water-filled pores between the cells. This process is known as paracellular transport, and is generally considered to be a passive process, even if this pathway appears to be selective for cationic rather than anionic and neutraldrugs. The paracellular pathway has also been shown to be saturable, by at least two separate mechanisms, one of which involves an intracellular process. Theparacellular permeability is dynamically regulated and varies both along the path of the intestine and along the crypt-villus axis. The average pore radius of the human small intestine is 8–13 Å, which will limit the paracellular permeability of drugs >4Å and restrict those >15 Å. The colon is even more size-discriminating since the paracellular pathway restricts drugs <3.5Å10.
Physicochemical properties of Permeation Enhancers:
Factors such as the log P, hydrogen bonding, solute charge, electrostatics of lipid bilayers, solute size and order of the partition phase describe and influence drug partitioning into cell membranes. The permeability of the drugs varies as function of surface area to volume ratio and regional pH effects on ionisation (Mälkiä et al., 2004). Chapter 4: Small intestine mucosa 135The driving force for diffusion across the apical and basolateral membranes of the enterocytes is the soluble drug concentration gradient and for ionisable drugs this varies with the pKa, the pH and the pH profile between the intestinal compartments (Hendriksen et al., 2003).
Permeability Enhancement Techniques:
Permeation enhancers
Protein-like compounds are generally not well absorbed through mucosae, due to their molecular size, hydrophilicity and metabolism occurring at the site of absorption. Permeation enhancers and/or delivery vehicles can also be used to enhance membrane transport of proteins, peptides and other chemical compounds across biological membranes (Veuillez et al., 2001).Permeation enhancers promote the transport of drugs through a mucosal membrane.Several classes of enhancers have been evaluated for potential use in enhancingtransmucosal delivery. It is clear from the literature that the choice of a permeation enhancer must be based on the following characteristics: effectiveness, safety, chemical inertness, lack of biological activity and rapidly reversibility of toxic and/orpharmacological effects, if any.Examples of strategies to improve peptide absorption include the following: chemicalenhancers, enzyme inhibitors, lipophilicity modification and formulation design (Veuillezet al., 2001).
Chemical enhancers
Examples of chemical enhancers often studied or used are surfactants, bile salts, chelators, alcohols and fatty acids. Chemical enhancers may exert their effect by:
1. altering the rheological properties of the mucus layer
2. increasing the thermodynamic properties of the peptide
3. enhancing transcellular transport by interacting with phospholipids and/or proteins to increase membrane fluidity
4. enhancing paracellular transport
5. inhibiting enzyme activity
(Veuillez et al., 2001)
* Surfactant
Currently much research involves studying the diffusion of small peptide molecules through biological membranes in the presence of chemical permeation enhancers, such as surfactant. Surfactants are used as emulsifier and as physical stabilising, wetting and suspending agents in many topical pharmaceutical formulations, cosmetic and food products. Surfactants may enhance partitioning by reducing the surface tension between the vehicle and the membrane surface and by influencing the barrier potential of the membrane and the tight junctions. It may also disrupt the barrier layers of the membrane (Fig. 6) (Barry and El Eini, 1976). Furthermore, the use of surfactants or complexation agents is associated with a reduction in the thermodynamic activity of the drug due to micellar association or complexation. In turn, this leads to changes in the free concentration of drug available for transport or diffusion across the membrane (Poelma et al., 1991).
* Bile salts
Bile, which contains glycine and taurine conjugates of cholic acid and chenideoxycholic acid, emulsifiesdietary fat and accelerates lipolysis and transport of lipid products through the unstirred water layer of the intestinal mucosa by micellar solubilisation. The bile salts which escape from active reabsorption in the ileum are metebolised to secondary bile salts deoxycholic acid & lithocholic acid by the bacterial flora. The diminishing order of hydrophilicity is as follows taurine conjugates > glycine conjugates > free bile salts. Polarity increases with the number of hydroxyl groups. Bile salts are capable to bind calcium, their binding properties decreasing with increasing hydrophilicity. No unambiguous data is available on the mechanism of absorption enhancement by bile salts. It may be carried out by effects on the mucous layer and on paracellular and transcellular absorption routes. They have been reported to affect the intestinal glycocalyx structure and to diminish gastric and intestinal mucous. A transcellular absorption enhancing effect is suggested by the phospholipid disordering action of unconjugated and conjugated bile salts. Colonic tight junction structure appears to be influenced by comparatively low bile salt concentrations (5mM and lower) in rabbits and rats. This paracellular absorption promoting effect is suggested to be intermediated by binding of Ca2+. Although bile salts have been validated to enhance drug uptake to a significantextent, applicability of these compounds as safe absorption promoter in man faces many complications, because mucosal damage seems to be correlated with their uptake .On the other hand, 2 year therapy with oral chenodeoxycholic acid (350-750 mg/day) for dissolution of gallstones was concomitant with mild side effects (increase of serum level of amino transferase and cholesterol, diarrhoea). This observation designates that longterm therapy with bile salts containing formulations may be promising in man. However, the suggested co-carcinogenic & co-mutagenic effects of secondary bile salts discourage the development of bile salts containing pharmaceutical formulations. (Ewoud et al1989).
* Straight chain fatty acids
The medium chain fatty acids including capric acid (C10), lauric acid (C12) and long chain fatty acids, such as oleic acid (C18) have been shown to increase the permeability of a series of hydrophilic drugs by dilating the tight junction and/or changing the cytoskeleton of the intestinal epithelial cells without prominent cytotoxicity. One of the foremost advantages of these excipients is the comfort of incorporating into the conventional oral dosage forms without the need for complex or expensive formulation technique. (Dimple et al2010)
1.Phosphatidyl-inositol-(4,5)diphosphate(PIP2) Ionosito1,4,5triphosphate(IP2)
2.Calmudin-dependent kinase(CaMK) Ca+2(release from endoplasmic reticulum)
3.Myosin light chain kinase Myosin light chain
4.Contractions in perijunctional actin-myosin ring
5.Increases the absorption of drug
* Chitosan derivatives
Chitosan is a non-toxic, biocompatible polymer that has found a number of applications in drug delivery including that of absorption of hydrophilic macromolecular drugs. Chitosan, when protonated (pH 6.5), is capable to increase the paracellular permeability of peptide drugs across mucosal epithelia. Chitosan derivatives have been assessed to overcome chitosan’s incomplete solubility and effectiveness as absorption enhancer at neutral pH values such as those found in the intestinal tract. Trimethyl chitosan chloride (TMC) has been synthesized at different degrees of quarterisation. This quaternized polymer forms complexes with anionic macromolecules and gels or solutions with cationic or neutral compounds in aqueous environment and neutral pH values. TMC has been shown to substantially increase the permeation and /or absorption of neutral and cationic peptide analogue across intestinal epithelia. The mechanism by which TMC enhances intestinal permeability is similar to that of protonated chitosan. It reversibly interacts with components of tight junctions, leading to widening of the paracellular routes. Monocarboxy methylated chitosan (MCC) is poly ampholytic polymer able to form visco-elastic gels in aqueous environments or with anionic macromolecules at neutral pH values. MCC appears to be less potent compared to the quaternized derivative. Nevertheless MCC is found to increase the permeation and absorption of low molecular weight heparin (LMWH; an anionic polysaccharide) across intestinal epithelia. Neither chitosan derivatives aggravates damage of the cell membrane, nor therefore do they not alter the variability of intestinal epithelia cells. (Thanou et al2001).
* Saponins
Saponins are glycosides of vegetable source with surface tension reducing properties and haemolytic action. They are capable of precipitating sterols and exert intestinal and transdermal absorption promoting properties. It is imaginable that the absorption supporting properties of saponins are mediated by their surfactants like properties. On the other hand, a transcellular promoting effect may also be caused by interaction with the membrane stabilizer cholesterol. This shows that saponins exhibit absorption promoting activity at relatively low concentrations. However; also for these compounds the issue of safety vs. efficacy requires further investigation. (Ewoud et al1989).
* Chelating agents:
Chelating agent forms complexation of calcium and magnesium ions present in between intestinal epithelial cells and ultimately leads to opening of tight junctions and thereby increasing permeability for exogenous substances.
* Zonula occludens toxin (Zot):
Zonula occludens toxin (Zot), a protein elaborated by Vibrio cholera that is able to reversibly regulate tight junction permeability. This toxin interacts with a specific intestinal epithelial surface receptor, with subsequent activation of a complex intracellular cascade of events that regulate tight junction permeability. It was also shown that the invitro permeabilities of drugs with low oral bioavailability such as paclitaxel, acyclovir, and cyclosporine and enamione anticonvulsants were increased with Zot.
* Polycarbophyl-cysteine conjugate(PCPCys):
It is a class of permeation enhancers is represented by thiolated polymers12 also called thiomers. These are polymers in which the thiol groups are covalently bound. It has been shown that polycarbophyl polymers (PCP) display permeation enhancing effects. This property is significantly improved as a result of the covalent attachment of cysteine (Cys) to this polymer (PCP-Cys). This thiolated polymer (PCP-Cys) is able to significantly increase the transport of marker compounds (sodium fluorescein) and peptide drugs (bacitracin-fluorescein isothiocyanate and insulin-fluoresceinisothiocyanate) across the intestinal mucosa of guinea pigs. The thiol groups,covalently attached to the polymer, seem to be responsible for the improved permeation-enhancing properties of these conjugates.These compounds exert their permeation enhancing effects via glutathione. It seems that PCP-cys can transform oxidized glutathione (GSSG) to reduced glutathione (GSH), prolonging GSH concentration at the apical membrane. GSH is reportedly capable of inhibiting protein tyrosine phosphatase (PTP) activity by almost 100%.
Vehicles and adjuvants (co-solvent)
A drug can be dissolved or dispersed in a solvent to improve transport by changing the thermodynamic activity and/or increasing the drugs solubility in the epithelial barrier of the mucosa (Veuillez et al., 2001).
Enzyme inhibitors:
Peptidase inhibitors may be employed to overcome both enzymatic barriers to permeation. It may be used alone or in combination with other permeation enhancers (Aungst, 1994).
Lipophilicity modification
The lipophilicity plays an important role in the diffusion of a peptide through a biological membrane. Acylation or alkylation may be used to conjugate the N-terminal with lipophilic molecules or prodrugs may be used to enhance lipophilicity and the diffusion of peptides (Veuillez et al., 2001).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Methods For Studying Drug Permeability:
In vivo methods
In vivo methods for evaluation of drug absorption are commonly used, even though it is extremely time and resource intensive due to experimental animal work and bio analysis of a large number of samples. It is based on the similarity of the composition of epithelial cell membranes of mammals and the fact that absorption is essentially an interaction between the drug and the biological membrane. These studies should be performed with caution due to potential species differences that may affect the drug absorption such as pH, GI motility, transit time and differential distribution of enzymes and transporters. Comparison of AUC (plasma concentration vs. time curve) values after intravenous, oral and intraportal (or intraperitoneal) administration can often indicate the absolute extent of in vivo drug absorption. This method is able to differentiate well absorbed compounds from reduced bioavailable compounds (Balimane et al., 2000).
In Silico methods
In silico methods are based on computational or virtual screenings of physico-chemical properties (such as lipophilicity, H bonding capacity, molecular size, polar surface area (PSA) and quantum properties) of drugs to predict their oral absorption potential. A variety of computational models exists, due to the increase in computer resources over the past decades. The in silico predictive models minimise the time consuming steps of synthesis and safe time by enabling screening of drug candidates prior to synthesis. These methods are very attractive, since intestinal absorption can be predicted before the synthesis of compounds, but at present their predictive power is unsatisfactory compared to experimental models (Corti et al., 2006; Balimane et al., 2000).To develop reliable and valid computational methods it is important that the quantitative structure-activity relationship (QSPR) models should be based on experimental data thatwere obtained from a diverse set of compounds with respect to their properties (e.g. physico chemical and pharmacological) and chemical structures. At present, most reports involve in silico modelling studies performed on compounds closely related in structure, thus making the model ineffective when applied to a wider structurally and diverse data set (Balimane et al., 2000). Lipinski and co-workers (2001) developed one of the best-known computational models to predict intestinal absorption form molecular properties. It is based on the physicochemical properties of 2200 drug candidates that had entered clinical phase II trials. They formulated a simple rule, taking into account hydrogen boding, molecular weight and lipophilicity. This so-called rule-of-five (ROF) states that a compound is likely to have limited oral bioavailability due to low intestinal epithelial permeability or solubility if it generates two or more alerts. The ROF is produced if the number of H-bond donors or acceptors in a compound exceeds 5 and 10 respectively; the molecular weight exceeds 500, or the lipophilicity expressed as the logarithm of the octanol-water partition coefficient (log P) exceeds 5. Weaknesses of the ROF include that it does not apply to compounds that are subject to active transport processes. It is mainly built on compounds with a high intestinal absorption and it gives relatively rough and sometimes false predictions (Bohets et al., 2001; Lipinski et al., 2001). Absorption is a very complex process that cannot be estimated accurately from structure alone. Several companies have therefore attempted to combine parameters that affect intestinal absorption in commercial available packages (Bohets et al., 2001).
* In vitro methods
For reasons of safety and cost, drug absorption studies (Sawada T et al ,1365–1371.) in humans are only carried out for a limited number of well characterized drugs. Studies of drug absorption in the intestine traditionally been carried out in experimental animals. However, the introduction of combinatorial chemistry and high throughput pharmacological screening in drug discovery has significantly increased the number of compounds entering thepre-clinical phase, and this has impossible to assess the absorption properties of all these compounds in experimental animals. This fact has spurred the development and use of vitro methods to assess drug permeability properties in most drug discovery settings. Also, the insight that drug absorption across biological barriers is a complex process involving several pathways that properties in most drug discovery settings. Also, the insight that drug absorption across biological barriers is a complex process involving several pathways that cannot easily be delineated in experimental animals has resulted in the large interest in academic and industrial institutions in these methods .The methods are, cultured cells and artificial membranes. Good oral availability is dependent on maximum intestinal permeability and maximum solubility/dissolution of the drug at the site of absorption. The extent of in vivo absorption can therefore be predicted based on in vitro permeability and solubility measurements. The research of intestinal permeability represents an essential part in the prediction of the oral bioavailability of any new drug candidate (Trapani et al., 2004). Due to the multiple processes involved in drug absorption in the intestine, it is often difficult to use just one in vitro model to accurately predict the in vivo permeability characteristics (Balimane et al., 2000). Different in vitro methods exist to assess the intestinal permeability of potential drug candidates. Each in vitro method has its distinct advantages and disadvantages. General advantages of in vitro techniques for assessment of permeability are that it is less labour and cost-intensive compared to in vivo animal studies. The successful application of in vitro models to predict drug absorption across the intestinal mucosa depends on how closely the in vitro model mimics the in vivo intestinal epithelium characteristics. Since it is very difficult to develop a single in vitro system that can simulate all the conditions existing in the human intestine, various in vitro systems are presently used as decision-making tools in early drug discovery.
* Animal tissue
Excised animal tissue models have been used since the 1950s to explore the mechanism of absorption of nutrients from the intestine. Animal intestinal tissues consist of essentially the same kind of endothelial cells than humans. Various animal spec








