
 About Authors:
About Authors:
Jatin Patel*, Krunal Parikh, Dhiren Shah
Seth G.L. Bihani S.D. College of Technical Education,
Institute of Pharmaceutical Sciences and Drug Research,
Sri Ganganagar, Rajasthan, INDIA
*Patelj313@yahoo.com
ABSTRACT:
A regulatory process, by which a person/organization/sponsor/innovator gets authorization to launch a drug in the market, is known as drug approval process. In general, a drug approval process comprises of various stages: application to conduct clinical trials, conducting clinical trials, application to marketing authorization of drug and post-marketing studies. Every country has its own regulatory authority, which is responsible to enforce the rules and regulations and issue the guidelines to regulate the marketing of the drugs.
This article includes new drug approval process in different countries include India, Australia, European union, China etc.
New drug approval process in different countries are described in logarithmic representation.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1531
INTRODUCTION
The new drug approval is of two phase process - the first phase for clinical trials and second phase for marketing authorization of drug. Firstly, non-clinical studies of a drug are completed to ensure efficacy and safety, and then application for conduct of clinical trials is submitted to the competent authority of the concerned country. Thereafter, the clinical trials can be conducted (phase I to phase IV). These studies are performed to ensure the efficacy, safety and optimizing the dose of drug in human beings. After the completion of clinical studies of the drug, then an application to the competent authority of the concerned country for the approval of drug for marketing is submitted. The competent authority review the application and approve the drug for marketing only if the drug is found to be safe and effective in human being or the drug have more desirable effect as compare to the adverse effect .
Even after the approval of new drug, government should monitor its safety due to appearance of some side effects, when it is used in larger population. The interactions with other drugs, which were not assessed in a pre-marketing research trial and its adverse effects (in particular populations) should also be monitored. [ Vyawahare.N.S, Itakar .S.C]
DRUG APPROVAL PROCESS IN OTHER COUNTRY
Drug Approval Process In USA
In 1820, the new era of USA drug regulation was started with the establishment of U.S. Pharmacopoeia. In 1906, Congress passed the original Food and Drugs Act, which require that drugs must meet official standards of strength and purity. However, in 1937, due to sulphanilamide tragedy, the Federal Food, Drug and Cosmetic Act (of 1938) was enacted and added new provisions that new drugs must be shown safe before marketing. Further, in 1962, the Kefauver-Harris Amendment Act was passed which require that manufacturers must prove that drug is safe and effective (for the claims made in labeling).
The Food and Drug Administration (FDA) is responsible for protecting and promoting public health. Like general drug approval process, FDA’s new drug approval process is also accomplished in two phases: clinical trials (CT) and new drug application (NDA) approval. FDA approval process begins only after submission of investigational new drug (IND) application. The IND application should provide high quality preclinical data to justify the testing of the drug in humans. Almost 85% of drugs are subjected to clinical trials, for which IND applications are filed. The next step is phase-I clinical trials (1-3 years) on human subjects (~100). The drug’s safety profile and pharmacokinetics of drug are focused in this phase. Phase II trials (2 years) are performed if the drug successfully passes phase I. To evaluate dosage, broad efficacy and additional safety in people (~300) are the main objective of the phase II. If evidence of effectiveness is shown in phase II, phase III studies (3-4 years) begins. These phase III concerns more about safety and effectiveness of drug from data of different populations, dosages and its combination with other drugs in several hundred to about 3,000 peoples. A new drug application (NDA) can be filed only when the drug successfully passes all three phases of clinical trials and includes all animal and human data, data analyses, pharmacokinetics of drug and its manufacturing and proposed labelling. The preclinical, clinical reports and risk-benefit analysis (product’s beneficial effects outweigh its possible harmful effects) are reviewed at the Center for Drug Evaluation and Research by a team of scientists. Generally approval of an NDA is granted within two years (on an average), however, this process can be completed from two months to several years. The innovating company is allowed to market the drug after the approval of an NDA and is considered to be in Phase IV trials. In this phase, new areas, uses or new populations, long-term effects, and how participants respond to different dosages are explored. Figure 1 represents the new drug approval process of FDA.
Drug Approval Process In Europe
In European Union (EU), the medical products were approved for marketing at the National level initially. The mutual recognization procedure was introduced in 1983 and a single national review in case of pharmaceutical/medicinal product for marketing authorizations in all EU’s countries was made feasible. The primary aim of this procedure was to create a united standard for product review among national regulatory authorities. In 1987, for high-technology or biologically derived products, the concertration procedure was established by directive 87/22, in which product assessment should be completed by Committee for Proprietary Medicinal Products (CPMP) besides the the normal national regulatory review. Further, in 1993, by council regulation (EEC) 2309/93, the concertration procedure was replaced with centralised procedure, by which all the high-tech and biologically derived product was reviewed and granted EU’s wide marketing authorization by the EU’s CPMP.
Similarly, the drug approval process in European countries is also accomplished in two phases: clinical trial and marketing authorization. A clinical trial application (CTA) is filed to the competent authority of the state to conduct the clinical trial within EU. The competent authority of that member state evaluates the application. The clinical trials are conducted only after the approval. The purpose and phases of clinical trials are similar as specified in FDA drug approval process. Figure 2 represent the clinical trial authorization process in EU.
After completing of all three phases of clinical trial, marketing authorization application is filed including all animal and human data, its analyses, as well as pharmacokinetics, manufacturing and proposed labelling. In the EU’s countries, the company have a choice of following regulatory procedures:
Centralized Procedure
The Committee for Human Medicinal Products (CHMP) evaluate the applications received by the EMEA. In view of the applicant's preference, CHMP contracts out assessment work in one of the member states (the "rapporteur"). After the complete assessment, the CHMP deliver opinion to EU Commission within 210 days. The EU Commission requests comments from other member states, if a positive opinion from CHMP is received. The other member states can respond in about 28 days. When a licence is recommended, a European Public Assessment Report (EPAR) is produced and marketing authorisation is issued. This authorisation is valid throughout the European Union and is for five years, however, the extension can be applied to the EMEA three months before the expiration of this period. Figure 3 represent the centralized procedure for marketing authorization.
Decentralised Procedure
In order to obtain marketing authorizations in several member states, the centralised procedure is not mandatory; in such case the decentralized procedure is to be used. An application is submitted to competent authorities of each of the member states, where a marketing authorization is to be sought. The information like quality, efficacy, safety, administrative information shall be submitted and a list of all Concerned Member States (CMSs) and one member state to act as Reference Member State (RMS). A draft assessment report on the medicinal product is prepared and the CMSs and the RMS validate the application within a time frame of 14 days. The RMS prepare draft summary of product characteristics, labeling and package leaflet within 120 days. This report can be approved within 90 days. However, if a medicinal product is supposed to cause potential serious risk to public health, CMS(s) will inform to other CMS, RMS and applicant and further decision in this regard is taken within 30 days. Within 60 days of the communication of the points of disagreement, all member states reach to an agreement on the action to be taken. After reaching to an agreement of the member states, the RMS records the agreement and informs to the applicant. However, if the member states could not reach an agreement, then CHMP intervenes and take a final decision keeping in view of the written or oral explanations of the applicant. Figure 4 represent the decentralized procedure for marketing authorization in EU.
National Procedure
This type of authorization is granted on country-by-country basis by the competent authorities, in each member state. Products only intended for one market and not obliged to use the centralized procedure.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Mutual Recognition Procedure
The mutual recognition procedure (MRP) is similar to the de-centralized procedure with some differences. The mutual recognition procedure is applicable to medicinal products which have received a marketing authorization in any member state whereas the decentralized procedure is applicable to those products which were never approved in any member states of the European Union. The MRP is used to obtain marketing authorizations in various several member states. The evaluation of application by RMS can be taken within 90 days instead of 120 days (in decentralized procedure). After the grant of marketing authorization, the product can be marketed, which may be called as Phase IV trials, wherein new uses or new populations, long-term effects etc. can be explored.
Drug Approval Process In China
In 1963, for the management of new drugs, Chinese Ministry of Health planned drug regulation. The China’s State Pharmaceutical Administration in collaboration with Ministry of Health, in 1979, published the New Drug Management Regulations (no need to conduct systematic scientific experiments on new drugs). In view of protecting the public health and promoting the economic developments in pharmaceuticals, the first comprehensive Drug Administrative Law was framed in 1985. This law was amended in 1999 by two additional provisions for new drug approval and provisions for new biological product approval. The approval process of New Drug Applications (NDA) includes sufficient preclinical data for verification of drug’s safety and justification of the commencement of clinical trials. The Drug Administrative Law was further revised in 2001 requiring premarket testing, approval for new drug products, and prohibits drug adulteration.
The Drug Administrative Law authorizes the State Food and Drug Administration (SFDA) to approve new drugs for marketing. The new drug registration process also consists of the clinical study application and the new drug application. The Provincial Drug Administration Authorities (PDAAs) should organize the works of the formal review of submitted materials i.e. on-site examination and sampling just after receiving the drug registration application. The aim behind the formal review is to guarantee the content and format of the submitted materials is in line with the requirements and all the required materials have been submitted. After formal review, the PDAAs send the qualified applications to the SFDA for further review. The import drug registration application should be directly submitted to SFDA by the applicant. SFDA’s Department of Drug Registration carefully reviews the completeness of the submitted materials, files the qualified applications and transmits all the materials of qualified applications to the Center for Drug Evaluation (CDE) directly attracted to SFDA. CDE determine whether the safety and effectiveness information submitted for a new drug are adequate for manufacturing and marketing approval and send the report of review to SFDA. SFDA Carefully consider the recommendations and review results of CDE and makes a decision whether or not the drug registration application can be approved and issues the certificate of drug approval and drug approval number to the qualified applicant. Figure 5 -6 represents the clinical trial approval process and new drug approval process of China, respectively.
Drug Approval Process In Australia
In the history of drug regulatory system in Australian, thalidomide disaster was a key factor. In 1948, the first advisory committee to review drugs was set up and further in 1964, the first Commonwealth advisory committee in Australia was established. The first federal act relating to therapeutic goods was enacted in 1965. In response of lacking control over locally manufactured products, the Therapeutic Goods Act was changed in 1989 and the Therapeutic Goods Administration (TGA) was created.
Any person seeking approval of a new drug in Australia should first file a clinical trial application for conducting human studies. The clinical trials in Australia can be conducted under two schemes, i.e. either under the Clinical Trial Exemption (CTX) Scheme or under the Clinical Trial Notification (CTN) Scheme. In the latter scheme, application is directly submitted to the Human Research Ethics Committee (HREC) which assesses the validity of design of clinical trial, its ethical acceptability, approval, safety and efficacy of the drug as well. The final consent for conducting trial is given by the approving authority after due advice from the HREC. The commencement of clinical trial takes place only after the due notification to the TGA and the appropriate notification fee to be paid. In CTX scheme, an application to conduct clinical trials is submitted to the TGA for evaluation and comment. The clinical trials can be conducted (under the CTX application) without further assessment by the TGA and the conduct of each trial should be notified to the TGA. Figure 7 represents the CTA by CTX scheme. An application is submitted to TGA to register the drug in Australian Register of Therapeutic Goods (ARTG) after the completion of clinical trials. The application consists of data to support the quality, safety and efficacy of the product for its intended use. The application is assessed (on an administrative level) to make sure for compliance with basic guidelines and further evaluated by different sections and advice can also be sought on key issues to take final decision. A company can make comments on the evaluation report, if necessary. A delegate (decision-maker) within the TGA after due advice of the ADEC, take a decision to approve or reject the product. The Australian Drug Evaluation Committee (ADEC) usually gives advice for new medicines. Figure 8 represent the new drug approval process in Australia. When the drug is approved and distributed in the market the drug, it is considered to be in Phase IV trials. In this phase, new uses or new populations, long-term effects, etc. can be explored.
Table 1. Comparison of Drug approval process.
|
Country |
Time for Regulatory Approval of CTA/IND Application |
Time for Evaluation of MAA |
MAA Fee |
|
Australia |
120 day |
50 days |
$192,400 |
|
China |
50 days |
180 days |
DNA |
|
India |
16-18 weeks |
8-12 weeks |
50,000 INR |
|
UK* |
35 days |
210 days |
£254100 |
|
USA |
30 days |
180 days |
$217,787 |
*By Centralized Procedure; MAA-Marketing Authorization Application, IND-Investigational New Drug, CTA-Clinical Trial Authorization, DNA-Data Not Available.
LOGARITHAMIC PRESENTATION OF NDA PROCESS IN DIFFERENT COUNTRIES:
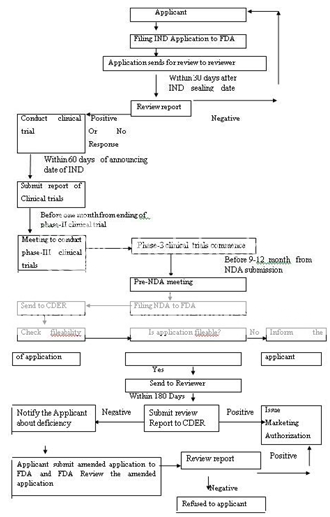
IND-Investigational New Drug, FDA-Food and Drug Administration, NDA-New Drug Application, CDER-Centre for Drug Evaluation and Research
Figure 1: New Drug Application Approval Process of FDA
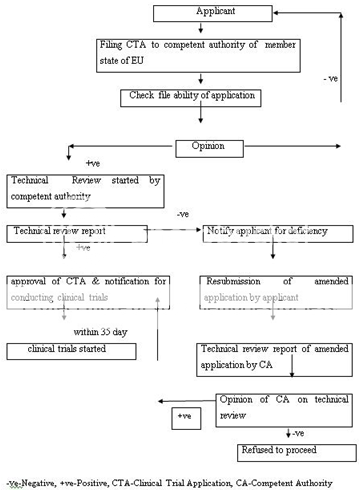
Figure 2: Clinical Trial Authorization Process of EU
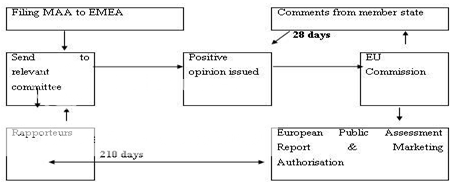
MAA-Marketing Authorization Application, EMEA-European Medicine Evaluation Agency, EU-European Unionion in EU
Figure 3:Centralized Procedure for Marketing Authorization
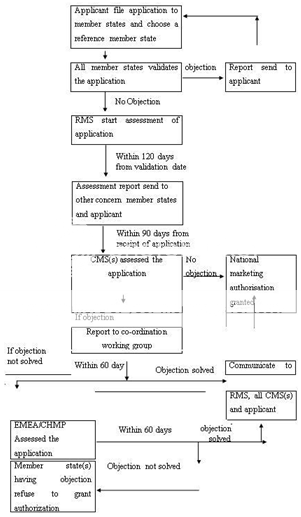
CMS(s)-Concerned Member State(s), RMS-Reference Member State, CHMP-Committee for Human Medicinal Products
Figure 4: Decentralised Procedure for Marketing Authorization in EU
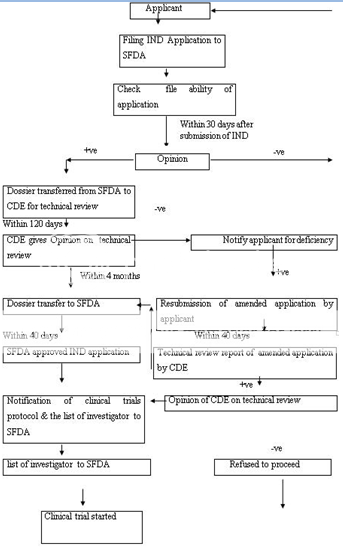
CDE-Centre for Drug Evaluation, SFDA-State Food and Drug Administration
Figure 5: Clinical Trial Application Approval Process of China
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
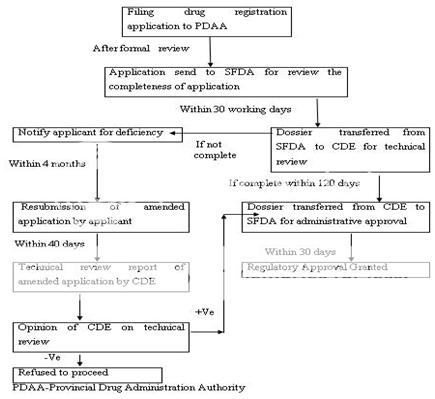
Figure 6: New Drug Registration Process of China
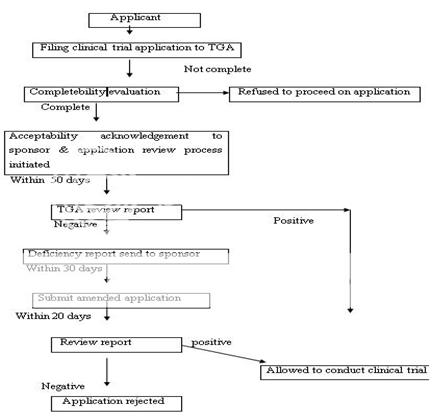
Figure 7: Clinical Trial Authorization Process of Australia under CTX Scheme
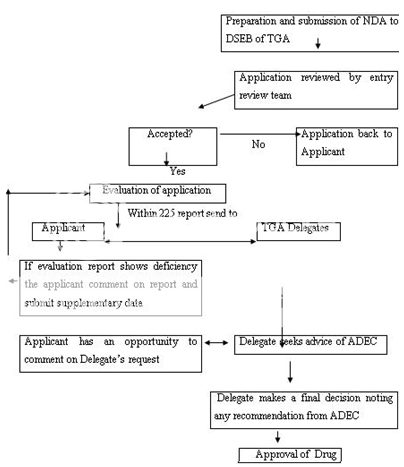
NDA-New Drug Application, DSEB-Drug Safety and Evaluation Branch, TGA-Therapeutic Goods Administration, ADEC-Australian Drug Evaluation Committee
Figure 8: New Drug Registration Process of Australia
[New%20Drug%20Approval%20Process%20%20Regulatory%20View%20%20%20Pharmainfo.net.htm]
DRUG APPROVAL PROCESS IN INDIA
1)investigation of new drug in india
Introduction
Current Federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines. Because a sponsor will probably want to ship the investigational drug to clinical investigators in many states, it must seek an exemption from that legal requirement. The IND is the means through which the sponsor technically obtains this exemption from the FDA.
During a new drug's early preclinical development, the sponsor's primary goal is to determine if the product is reasonably safe for initial use in humans, and if the compound exhibits pharmacological activity that justifies commercial development. When a product is identified as a viable candidate for further development, the sponsor then focuses on collecting the data and information necessary to establish that the product will not expose humans to unreasonable risks when used in limited, early-stage clinical studies.
FDA's role in the development of a new drug begins when the drug's sponsor (usually the manufacturer or potential marketer) having screened the new molecule for pharmacological activity and acute toxicity potential in animals, wants to test its diagnostic or therapeutic potential in humans. At that point, the molecule changes in legal status under the Federal Food, Drug, and Cosmetic Act and becomes a new drug subject to specific requirements of the drug regulatory system.
The IND application must contain information in three broad areas:
· Animal Pharmacology and Toxicology Studies - Preclinical data to permit an assessment as to whether the product is reasonably safe for initial testing in humans. Also included are any previous experience with the drug in humans (often foreign use).
· Manufacturing Information - Information pertaining to the composition, manufacturer, stability, and controls used for manufacturing the drug substance and the drug product. This information is assessed to ensure that the company can adequately produce and supply consistent batches of the drug.
· Clinical Protocols and Investigator Information - Detailed protocols for proposed clinical studies to assess whether the initial-phase trials will expose subjects to unnecessary risks. Also, information on the qualifications of clinical investigators--professionals (generally physicians) who oversee the administration of the experimental compound--to assess whether they are qualified to fulfill their clinical trial duties. Finally, commitments to obtain informed consent from the research subjects, to obtain review of the study by an institutional review board (IRB), and to adhere to the investigational new drug regulations.
Once the IND is submitted, the sponsor must wait 30 calendar days before initiating any clinical trials. During this time, FDA has an opportunity to review the IND for safety to assure that research subjects will not be subjected to unreasonable risk.
[Investigational%20New%20Drug%20(IND)%20Application.htm]
2) procedure for new drug approval in india
The Drug and Cosmetic Act 1940 and Rules 1945 were passed by the India's parliament to regulate the import, manufacture, distribution and sale of drugs and cosmetics. The Central Drugs Standard Control Organization (CDSCO), and the office of its leader, the Drugs Controller General (India) [DCGI] was established.
In 1988, the Indian government added Schedule Y to the Drug and Cosmetics Rules 1945. Schedule Y provides the guidelines and requirements for clinical trials, which was further revised in 2005 to bring it at par with internationally accepted procedure. The changes includes, establishing definitions for Phase I–IV trials and clear responsibilities for investigators and sponsors.
The clinical trials were further divided into two categories in 2006. In one category (category A) clinical trials can be conducted in other markets with competent and mature regulatory systems whereas the remaining ones fall in to another category (category B) Other than A.
Clinical trials of category A (approved in the U.S., Britain, Switzerland, Australia, Canada, Germany, South Africa, Japan and European Union) are eligible for fast tracking in India, and are likely to be approved within eight weeks. The clinical trials of category B are under more scrutiny, and approve within 16 to 18 weeks.
An application to conduct clinical trials in India should be submitted along with the data of chemistry, manufacturing, control and animal studies to DCGI. The date regarding the trial protocol, investigator's brochures, and informed consent documents should also be attached. A copy of the application must be submitted to the ethical committee and the clinical trials are conducted only after approval of DCGI and ethical committee. To determine the maximum tolerated dose in humans, adverse reactions, etc.
On healthy human volunteers, Phase I clinical trials are conducted. The therapeutic uses and effective dose ranges are determined in Phase II trials in 10-12 patients at each dose level. The confirmatory trials (Phase III) are conducted to generate data regarding the efficacy and safety of the drug in ~ 100 patients (in 3-4 centers) to confirm efficacy and safety claims. Phase III trials should be conducted on a minimum of 500 patients spread across 10-15 centers, If the new drug substance is not marketed in any other country.
The new drug registration (using form # 44 along with full pre-clinical and clinical testing information) is applied after the completion of clinical trials. The comprehensive information on the marketing status of the drug in other countries is also required other than the information on safety and efficacy. The information regarding the prescription, samples and testing protocols, product monograph, labels, and cartons must also be submitted.
The application can be reviewed in a range of about 12-18 months. Figure 10 represents the new drug approval process of India. After the NDA approval, when a company is allowed to distribute and market the product, it is considered to be in Phase IV trials, in which new uses or new populations, long-term effects, etc. are explored.
The drug approval process varies from one country to another. In some countries, only a single body regulates the drugs and responsible for all regulatory task such as approval of new drugs, providing license for manufacturing and inspection of manufacturing plants e.g. in USA, FDA performs all the functions. However in some counties all tasks are not performed by a single regulatory authority, such as in India, this responsibility is divided on Centralised and State authorities. Other issues where the difference appears are, time taken for the approval of a CTA application, time taken in evaluation of marketing authorization application, registration fee, registration process and marketing exclusivity .
Some counties have two review processes as normal review process and accelerated review process as in USA, China etc. and some countries have only a single review process as in India. Similarly, the format used for the presentation of dossier submitted for approval of drug is also different. In some countries like as in USA, EU, and Japan , it is mandatory that the dossier prepared in CTD format, however, in some countries it is optional such as in India.
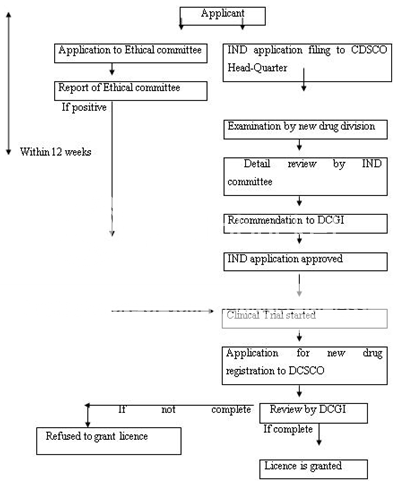
IND-Investigational New Drug, DCGI-Drug Controller General of India, CDSCO-Centre for Drug Standards Control organization.
Figure 9: New Drug Registration Process of India
[New%20Drug%20Approval%20Process%20%20Regulatory%20View%20%20%20Pharmainfo.net.htm]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
CTD GUIDELINE IN INDIA:
Scope:
This guideline applies to import / manufacture and marketing approval of new drugs including New chemical entity, new indication, new dosage forms, modified release form, new route of administration etc. under the definition of new drug under Rule 122E of Drugs & Cosmetics rules as a finished pharmaceutical product.
What is CTD?
The CTD is only a format for submission of information to CDSCO. It does not define the content.
Difference in organization of data in each application has made reviewing more difficult and can also lead to omission of critical data or analysis so unnecessary delay in approval. Thus common format of submission will help. Through the ICH process, CTD guidance developed for japan,EU & US. CDSCO also adopted the CTD.
Guidelines for preparation of CTD
CTD: over view
Module 1: General Information
This module should contain documents specific to India; for example, Form 44, Treasury challan fee or the proposed label for use in India.
1) Covering letters & comprenshive table of contents (module 1 to 5)
2) Administrative information
Brief introduction about the applicant company
Duly filled and signed application form 44 and treasury challan
Legal and critical documents
Coordinates related to the application
General information of the drug product
Summary of the testing protocol(s) for quality control testing
Regulatory status in other countries
Domestic price of the drug followed in the countries of origin
Brief profile of manufacturer’s company & business activity
Information regarding involvement of expert if any
Samples of drug product
Promotional material
Module 2: CTD Summaries
This module should begin with a general introduction to the pharmaceutical, including its pharmacologic class, mode of action, and proposed clinical use, not exceeding one page. Module 2 should contain 7 sections in the following order:
1) CTD table of contents
2) CTD introduction
3) Quality overall summary
4) Nonclinical overview
5) Clinical overview
6) Nonclinical written and tabulated summaries
7) Clinical summary
In this module following information is required
1) Table of content of module
2) Introduction
3) Quality overall summary
In this section not provide the entire information it is presented in module 3. It is not more than 40 pages.
4) Summary of drug substance & drug product
5) Nonclinical overview
6) Clinical over view
MODULE:3 quality
In this module following information is required
1) Table of contents of module 3
2) Drug substances
3) Manufacture of drug substances
4) Characterization of drug substances
5) Quality control of drug substances
6) Reference standards and material
7) Container closer system
8) Stability of drug substance
9) Drug product and manufacture of drug product
10) Control of drug product and excipient
MODULE:4 non clinical study report
Table of contents in this module should be provided that lists all of the nonclinical study reports and gives the location of the each study reports in CTD.
It contains following data
1) study reports
Pharmacology
Pharmacokinetic
Toxicology
2) literature references
MODULE:5 clinical study report
It contains tabular listing of all clinical studies. Following data are required
1) clinical study report
Reports of biopharmaceutical studies
Reports of studies pertinent to pharmacokinetic using human biomaterials.
Reports of human pharmacokinetic studies
Reports of human pharmacodynamic studies
Reports of efficacy and safety study
Reports of post marketing experience
Case report form and individual patient listing
Literature references [CDSCO Guideline]
DOCUMENTS TO BE SUBMITTED FOR GRANT OF PERMISSION TO CONDUCT BIOEQUIVALENCE STUDIES FOR EXPORT PURPOSE:
A large number of applications are being filed to the office of DCG (I) at CDSCO (HQ) by Pharmaceutical companies, both manufacturers and importers as well as CRO’s on behalf of them, requesting for the approval to carry out BE studies with various pharmaceutical dosage formulations on Indian subjects.
In light of the above, for easy processing of such applications and to bring uniformity in decision making all stake holders of the afore mentioned activities are hereby advised to submit their applications with following documents. All applications should accompany the documents with proper index & page number.
New Drugs – developed in India as an IND and not marketed anywhere in world.
1. Form 44
2. Treasury Challan of INR 50,000.
3. Source of bulk drugs /raw materials.
Requirements for BE study of a new molecule not approved in India but approved in the other countries:
1. Application in Form-44 duly signed, by the competent authority with name and designation.
2. Treasury Challan of Rs. 50000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval granted to the BE study centre by CDSCO.
5. Sponsor’s Authorization letter duly signed by the competent authority on their letterhead.
6. The study protocols.
7. The study synopsis
8. Pre-clinical single dose data and repeated dose toxicity data.
9. Clinical study data and published report of pharmacokinetic and pharmacodynamic study carried out in healthy volunteers/patients data published in reputed journals.
10. Regulatory status of the drug.
11. Names of the countries where the drug is currently being marketed (to be mentioned in the covering letter also).
12. Package literature on the international product
13. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
14. In the case of multiple dose BE study adequate supporting safety data should be submitted.
15. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
16. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
New Drugs approved in India within period of 1 year:
1. Application in Form-44 duly signed, by the competent authority with name and designation
2. Treasury Challan of Rs. 25000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval of the BE study centre from CDSCO.
5. Sponsor’s Authorization letter duly signed by the competent authority on their letterhead.
6. The study protocols.
7. Clinical study data and published report of pharmacokinetic and pharmacodynamic study carried out in healthy volunteers data published in reputed journals.
8. Package literature on the international product.
9. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
10. In the case of multiple dose BE study adequate supporting safety data should be submitted.
11. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
12. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
New Drugs approved within period of more than 1 year & less than 4 years:
1. Application in Form-44 duly signed, by the competent authority with name and designation
2. Treasury Challan of Rs. 15000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval of the BE study centre from CDSCO.
5. Sponsor’s Authorization letter duly signed on their letterhead by the competent authority.
6. The study protocols.
7. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
8. In the case of multiple dose BE study adequate supporting safety data should be submitted.
9. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
10. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
BE NOC for all the drug products in modified release form irrespective of their approval status:
1. Application in Form-44 duly signed, by the competent authority with name and designation
2. Treasury Challan of Rs. 15000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval of the BE study centre from CDSCO.
5. Sponsor’s Authorization letter duly signed on their letterhead by the competent authority.
6. The study protocols.
7. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
8. In the case of multiple dose BE study adequate supporting safety data should be submitted.
9. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
10. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
All above requirements are general in nature, however depending on the nature of the drug, disease and studies further specific information may also be required to be furnished by the firm. [CDSCO guideline & Schedule-Y]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
APPLICATION FORM FORMAT:
Form 44(INDIA)
(See rules 122 A, 122 B, 122 D, and 122 DA)
Application for grant of permission to import or manufacture a New Drug or to undertake clinical trial.
*****
I/we____________________________________________ of M/s.
_____________________________________(address)
hereby apply for grant of permission for import of and/or clinical trial or for approval to manufacture a new drug or fixed dose combination or subsequent permission for already approved new drug. The necessary information / data is given below:
1.Particulars of New Drug:
(1) Name of the drug:
(2) Dosage Form:
(3) Composition of the formulation:
(4) Test specification:
(i) active ingredients:
(ii) inactive ingredients:
(5) Pharmacological classification of the drug:
(6) Indications for which proposed to be used:
(7) Manufacturer of the raw material (bulk drug substances):
(8) Patent status of the drug:
2.Data submitted along with the application (as per Schedule Y with indexing and page nos.)
A. Permission to market a new drug :-
(1) Chemical and Pharmaceutical information
(2) Animal Pharmacology
(3) Animal Toxicology
(4) Human/Clinical Pharmacology (Phase I)
(5) Exploratory Clinical Trials (Phase II)
(6) Confirmatory Clinical Trials (Phase III) (including published review articles)
(7) Bio-availability, dissolution and stability study Data
(8) Regulatory status in other countries
(9) Marketing information:
(a) Proposed product monograph
(b) Drafts of labels and cartons
(10) Application for test license
B. Subsequent approval / permission for manufacture of already approved new drug
(a) Formulation:
(1) Bio-availability/ bio-equivalence protocol
(2) Name of the investigator/center
(3) Source of raw material (bulk drug substances) and stability study data.
(b) Raw material (bulk drug substances)
(1) Manufacturing method
(2) Quality control parameters and/or analytical specification, stability report.
(3) Animal toxicity data
C. Approval / Permission for fixed dose combination:
(1) Therapeutic Justification (authentic literature in pre-reviewed journals/text books)
(2) Data on pharmacokinetics/pharmacodynamics combination
(3) Any other data generated by the applicant on the safety and efficacy of the combination.
D. Subsequent Approval or approval for new indication – new dosage form:
(1) Number and date of Approval/permission already granted.
(2) Therapeutic Justification for new claim / modified dosage form.
(3) Data generated on safety, efficacy and quality parameters.
A total fee of rupees_______________ (inwords).______________) has been credited to the Governmentunder the Head of Account________________________ (Photocopy of receipt isenclosed).
Dated_____
Signature__________________
Designation________________
Note- Delete, whichever is not applicable
[Form%2044.htm& Vyawahare.N.S, Itakar .S.C]


[Guarino R.A]
CONCLUSION
Generally, the drug approval process comprised mainly the two steps, application to conduct clinical trial and application to the regulatory authority for marketing authorization of drug. The new drug approval process of various countries is similar in some of the aspects whereas it differs in some aspects. In most of the counties, sponsor firstly files an application to conduct clinical trial, and only after the approval by the regulatory authority, the applicant conducts the clinical studies and further submits an application to the regulatory authority for marketing authorization of drug. In all countries, information submitted to regulatory authorities regarding the quality, safety and efficacy of drug is similar; however, the time, fee and review process of clinical trials and marketing authorization application differs.
For the purpose of harmonization, the International Conference on Harmonization (ICH) has taken major steps for recommendations in the uniform interpretation and application of technical guidelines and requirements. This step will ultimately reduce the need to duplicate work carried out during the research and development of new drugs. Therefore, harmonization of drug approval processes either by ICH or WHO may be initiated at global level.
BIBLIOGRAPHY:
- Central drugs standard control organization directorate general of health services ministry of health & family welfare govt. Of india, 24thaugust 2011.
- Form 44 format at URL Form%2044.htm accessed on 15/7/2011.
- Guarino, R.A. New drug approval process. Third edition. Marcel Dekker publication. New York. Pg.no-69-70
- Investigation of new drug by U.S. Food and drug administration at URL Investigational%20New%20Drug%20(IND)%20Application.htm accessed on 15/7/2011.
- New drug approval process in india by Dr. Harish Dureja at URL New%20Drug%20Approval%20Process%20%20Regulatory%20View%20%20%20Pharmainfo.net.htm accessed on 21/7/2011.
- Schedule y at URL Schedule%20Y(ammended%20version)%20-%20CDSCO.htm accessed on 15/7/2011.
- Vyawahare, N.S. Itakar, S.C. Drug regulatory affairs. Published by Nirali Prakashan. Pg.no-4.1, 16.







