
{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
Imran khan*, M. Idreesh khan, Unis khan
Dept. of Pharmaceutics in Shri Ram College of pharmacy, Banmore, Morena,
Madhya Pradesh, India
khan.imran731@gmail.com
ABSTRACT:
This is the novel approach for enhancing dissolution and bioavailability of BCS-II class drugs. Solubility is the major problem in the development of pharmaceutical dosage forms. Liquisolid technique is the novel and promising technique to overcome these problems. The liquisolid systems are to improve the dissolution properties of water insoluble agents. For enhancing the dissolution rates for water insoluble drugs we used carrier and coating materials like microcrystalline cellulose (Avicel), silica (Aerosil), sodium starch glycolate and magnesium stearate etc. These agents can significant effect on dissolution properties of water insoluble drugs.
REFERENCE ID: PHARMATUTOR-ART-2173
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 6 Received On: 30/03/2014; Accepted On: 04/04/2014; Published On: 01/06/2014 How to cite this article: I Khan, MI Khan, U Khan; Liquisolid Technology: An Emerging and Advance Technique for Enhancing Solubilization; PharmaTutor; 2014; 2(6); 31-41 |
INTRODUCTION:
Solubility is one of the important parameter to achieve the desired concentration of drug in systemic circulation for pharmacological response to be shown.1 two consecutive transport processes can be identified to describe the oral absorption of drugs from solid dosage forms.
(1) Dissolution of the drug in vivo to produce a solution
(2) Transport of the dissolved drug across the G.I. membrane.2
Poorly water soluble drugs are defined as those with high permeability but whose solubility in aqueous media is not sufficient for the whole dose to be dissolved in the gastrointestinal tract. For these substances dissolution is therefore the rate determining step to absorption. The poor dissolution rates of poorly water soluble drugs is still a substantial problem confronting drug development, such as hindering the development of parenteral products and limiting the bioavailability of oral products.3
The term “water-insoluble drugs” are the drugs which are known as-
Table: Different solubilities
|
Sparingly water-soluble |
1 part of solute to 100 parts of water |
|
Slightly water-soluble |
1 part of solute into 100 to 1000 parts of water |
|
Very slightly water soluble |
1 part of solute into 1000 to 10,000 parts of water |
Solubilization: -Solubilization is the process of bringing into solution substances which are insoluble or sparingly soluble in water.
Solubilization techniques in brief: there are various techniques available to improve the solubility of poorly soluble drugs. Some of the approaches to improve the solubility are:4
PH adjustment:
It is well documented that the influence of the changes in pH within the gastrointestinal tract upon the bioavailability of pharmaceuticals. The absorption of drug is largely dependent upon diffusion, which varies with pH of the individual regions within the gastrointestinal tract, the pKa of the drug and permeability, which are not only moderated by the surface area of the region in which it is released, but also the regional pH effects upon drug ionization. By applying a pH change, poorly water soluble drugs with parts of the molecule that can be protonated (base) or deprotonated (acid) may potentially be dissolved in water. While the importance of critical parameters like salt selection and pH adjustment has been stressed on pre-formulation, the use of pH altering excipients within drug delivery systems is also of significant utility. pH adjustment can in principle be used for both oral and parenteral administration.
Micro-emulsion:
A micro emulsion is an optically clear pre-concentrate, isotropic, thermo dynamically stable transparent (or translucent) system, containing a mixture of oil, hydrophilic surfactant and hydrophilic solvent which dissolves a poorly water soluble drug. Upon contact with water, the formulations spontaneously disperse (or ‘self emulsifies’) to form a very clear emulsion of exceedingly small and uniform oil droplets containing the solubilized poorly soluble drug. Micro-emulsions have been employed to increase the solubility of many drugs that are practically insoluble in water, along with incorporation of proteins for oral, parenteral, as well as percutaneous/transdermal use.
Self-emulsifying drug delivery systems:
Self-emulsifying or self-micro emulsifying systems use the concept of in situ formation of emulsion in the gastrointestinal tract. The mixture of oil, surfactant, co-surfactant, one or more hydrophilic solvents and co-solvent forms a transparent isotropic solution that is known as the self-emulsifying drug delivery system (SEDDS)
Manipulation of solid state:
From the stability and bioavailability aspects, the crystalline form of a drug is of pharmaceutical importance. Polymorphism (existence of a drug substance in multiple crystalline forms) can cause variations in melting point, density, stability and drug solubility as these properties depend on the escaping tendency of the molecules from a particular crystalline structure. As a rule, for a drug that have the highest order of crystallinity is the most stable form, exists in multiple polymorphic forms, i.e. with the least amount of free energy, and, consequently, possesses the highest melting point and the least solubility. By controlling the crystallization process, amorphous or Meta stable forms of drugs possessing high free energy can be forcibly created. They offer the advantage of higher solubility but suffer from stability issues unless stabilizers intended to inhibit crystal growth are incorporated in the formulation.
Particle size reduction:
The bioavailability of poorly soluble drugs is often intrinsically related to drug particle size. By reducing particle size, the increased surface area may improve the dissolution properties of the drug to allow a wider range of formulation approaches and delivery technologies.
Co-solvency:
The solubility of a poorly water soluble drug can be increased frequently by the addition of a water miscible solvent in which the drug has good solubility known as co-solvents. The use of co solvents is a highly effective technique to enhance the solubility of poorly soluble drugs.
Micellar solubilization:
The use of surfactants to improve the dissolution performance of poorly soluble drug products has also been successfully employed. Surfactants can lower surface tension and improve the dissolution of lipophilic drugs in aqueous medium. Commonly used non-ionic surfactants include polysorbates, polyoxyethylated castor oil, polyoxyethylated glycerides, lauroyl macroglycerides and mono- and di-fatty acid esters of low molecular weight polyethylene glycols.
Micelles are divided into 2 categories-
Mixed micelles:
Mixed micelles have a hydrophobic core in which low soluble compounds can dissolve. The micelle solubilized host molecules (i.e. drugs) in the micelle volume, but the penetration into the micelle depends over all on the inner space of the micelle, on the hydro-phobicity of the drug and on the charge of the incorporated molecule. The interaction between micelles and lipophilic drugs leads to the formation of mixed micelles (MM), often called swollen micelles, too.
Polymeric Micelles:
Amphiphilic polymers assemble into nano-scopic supra molecular core-shell structures, known as polymeric micelles, which are under extensive study for drug delivery. Polymeric micelles may be safe for parenteral administration relative to existing solublizing agents, permitting an increase in the dose of potent toxic and poorly water soluble compounds. Polymeric micelles solubilized important poorly water-soluble compounds.
Hydrotropy:
Hydrotropy is a solubilization process whereby addition of a large amount of second solute results in an increase in the aqueous solubility of another solute. Hydrotropy designate the increase in solubility in water due to the presence of large amount of additives.
Solid Dispersions:
In this technique, a poorly soluble drug is dispersed in a highly soluble solid hydrophilic matrix, which enhances the dissolution of the drug. Solid dispersion techniques can yield eutectic (non-molecular level mixing) or solid solution (molecular level mixing) products. Presence of the drug in microcrystalline state, improved wettability and formation of high free energy amorphous forms of the drug during solid dispersion formation contribute towards enhancement of drug solubilization.The most commonly used hydrophilic carriers for solid dispersions include polyvinylpyrrolidone, polyethylene glycols, Plasdone-S630, Tween-80, Docusate sodium, Myrj-52, Pluronic-F68 and Sodium Lauryl Sulphate used.
Nano-suspension:
This technology is applied to poorly soluble drugs that are insoluble in both water and oils. The particle size distribution of the solid particles in Nano-suspensions is usually less than one micron with an average particle size ranging between 200 and 600 nm.
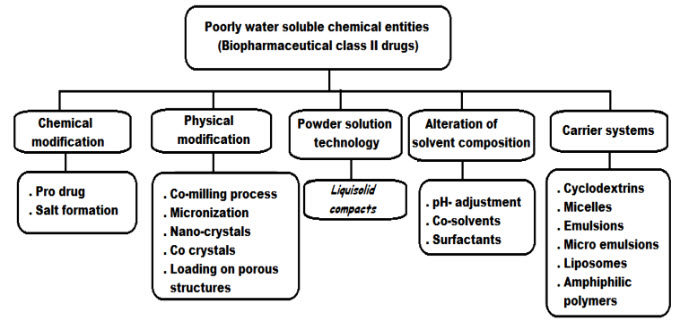
Fig: Different method for enhancing the solubility of drugs
Liquisolid Technology:
The concept of liquisolid compacts can be used to formulate liquid medication such as oily liquid drug and solutions or suspensions of water-insoluble solid drugs in non-volatile vehicles, into acceptably flowing and compressible powders.5
Using this new formulation technique, a liquid medication may be converted into a dry-looking, non-adherent, free flowing and readily compressible powder by a simple blending with selected powder excipients referred to as carrier and coating materials. Various grades of cellulose, starch, lactose, etc, may be used as the carrier, whereas a very fine particle size silica powder may be used as the coating material. Liquisolid compacts are acceptably flowing and compressible powdered forms of liquid medications and they are industrially applicable.
In addition, the term “liquid medication” does not only imply drug solutions, as in “powdered solutions”, but also drug suspensions, emulsions, or liquid oily drugs. Therefore, in contrast to “powdered solutions”, the term “liquisolid compacts” is more general and it may encompass for different formulation systems, namely, “powdered drug solutions”, “powdered drug suspensions”, “powdered drug emulsions”, and “powdered liquid drug”. Furthermore, the older term of “powdered solutions” seems to be inadequate even in describing the original systems, since it has not been proven that the remains in solutions in the liquid vehicle after its deposition on the extremely large powder surfaces of silica used.
Definition:
The various definition and terms which are used in liquisolid technology are:
Liquid medication: Includes liquid lipophilic drugs and drug suspensions or solutions of solid water insoluble drugs in suitable non-volatile solvent systems.
Liquisolid systems: Refers to powdered forms of liquid medications formulated by converting liquid lipophilic drugs or drug suspensions or solutions of water insoluble solid drugs in suitable nonvolatile solvent systems, into dry, non-adherent, free-flowing and readily compressible powder admixtures by blending with selected carrier and coating materials.
Carrier material: These are compression-enhancing, relatively large, preferably porous material possessing sufficient absorption properties which contributes in liquid absorption. e.g. Microcrystalline and amorphous cellulose,various grades of cellulose, starch, lactose, sorbitol, Avicel PH 102 and 200, Eudragit RL and RS, amorphous cellulose etc.6,7,8
Coating material: Refers to a material possessing fine and highly adsorptive particles, such as various types of silica, which contributes in covering the wet carrier particles and displaying a dry looking powder by adsorbing any excess liquid.These are flow-enhancing, very fine (10 nm to 5,000 nm in diameter) particles. e.g. silica of various grades like Cab-O-Sil M5, Aerosil 200, Syloid 244FP etc.
Non-volatile solvents: Inert, high boiling point, preferably water-miscible and not highly viscous organic solventsystems. Various non-volatile solvents used for the formulation of liquisolid systems includepolyethylene glycol 200 and 400, glycerin, polysorbate 80 and propylene glycol, liquid polyethylene glycol, polysorbate, glycerin, N, N dimethylacetamide, fixedoils etc.
Disintegrants: Disintegrants are those materials which are required to disintegrate the tablet after administration inside the body.Most commonly used Disintegrants is sodium starch glycolate (Explotab13, Pumogel etc.).
Preparation of liquisolid tablets:
The calculated quantities of drug and non-volatile liquid are accurately weighed in a glass beaker and then the beaker is sonicated for homogeneous mixing. The resulting medication is incorporated into other beaker Calculated quantities of carrier and coating materials are added and stirring is done completely. Later each selected liquisolid formula is blended with certain amount of superdisintegrant (used in formulations) and prepared liquisolid systems that are proved to have acceptable flowability and compressibility are compressed into tablets of desired weight using press with appropriate diameter of punch and die.9
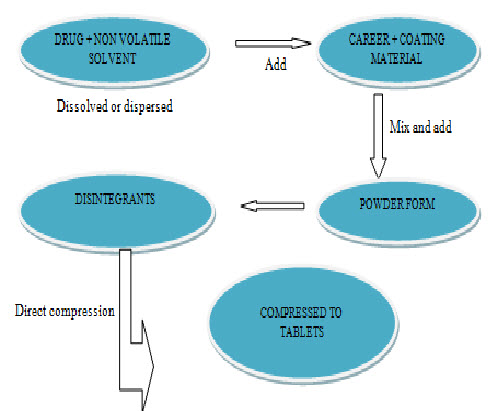
Fig: preparation of liquisolid tablets
Classification of liquisolid systems:
Based on the type of liquid medication contained therein, liquisolid systems may be classified into three sub-groups:
1)Powdered drug solutions
2)Powdered drug suspensions
3)Powdered liquid drugs
Powdered drug solutions and suspensions may be produced by the conversion of drug solutions or drug suspensions into liquisolid systems and powdered liquid drugs are produced from the formulation of liquid drugs into liquisolid systems. Simultaneously, based on the formulation technique used, liquisolid systems may be classified into two categories namely,
· Liquisolid compacts
· Liquisolid Microsystems
The term “liquisolid compacts” refers to immediate or sustained release tablets or capsules prepared, combined with the inclusion of appropriate adjuvants required for tabletting or encapsulation, such as lubricants, and for rapid or sustained release action, such as Disintegrants or binders, respectively.
The term “liquisolid Microsystems” refers to capsules prepared by combining the drug with carrier and coating materials, combined with inclusion of an additive e.g., PVP in the liquid medication wherein the resulting unit size may be as much as five times that of liquisolid compacts.
Wettability of liquisolid tablet:-
Wetting is the ability of a liquid to maintain contact with a solid surface, resulting from intermolecular interactions when the two are brought together. The degree of wetting (wettability) is determined by a force balance between adhesive and cohesive forces.The wettability of the compacts by the dissolution media is one of the proposed mechanisms for explaining the enhanced dissolution rate from the liquisolid compacts. Non-volatile solvent present in the liquisolid system facilitates wetting of drug particles by decreasing interfacial tension between dissolution medium and tablet surface.
Figure shows lower contact angle of liquisolid compacts than the conventional tablets and thus improved wettability.

Fig: Wettability of liquisolid tablet
Advantages:
1. Liquisolid systems are low cost formulations than soft gelatin capsules.
2. Drug release can be modified using suitable formulation ingredients
3. Drug can be molecularly dispersed in the formulation.
4. Industrial production is also possible.
5. Enhanced bioavailability can be obtained as compared to conventional tablets.
6. Several slightly, very slightly water-soluble, practically water-insoluble liquid and solid drugs can be formulated into liquisolid systems.
7. Even though the drug is in a tablet or capsule form, it is held in a solubilised liquid state, which contributes to increased drug wetting properties, thereby enhancing drug dissolution.
- Rapid release liquisolid tablets or capsules of water insoluble drugs exhibit enhanced in-vitro and in-vivo drug release when compared to their commercial counter parts, including soft gelatincapsules preparation.
- Sustained release liquisolid tablets or capsules of water insoluble drugs exhibit constant dissolution rates (zero-order release) comparable only to expensive commercial preparations that combine osmotic pump technology and laser-drilled tablets.10,
- Can be applied to formulate liquid medications such as oily liquid drugs.
- Better availability of an orally administered water insoluble drug.
- Production of liquisolid systems is similar to that of conventional tablets.
- Can be used for formulation of liquid oily drugs.
- Can be used in controlled drug delivery.10
Limitations:
1. Not applicable for formulation of high dose insoluble drugs.
2. If more amount of carrier is added to produce free-flowing powder, the tablet weight increases to more than one gram which is difficult to swallow.
3. Acceptable compression properties may not be achieved since during compression liquid drug may be squeezed out of the liquisolid tablet resulting in tablets of unsatisfactory hardness.
4. Introduction of this method on industrial scale and to overcome the problems of mixing small quantities of viscous liquid solutions onto large amounts of carrier material may not be feasible.
Applications:
Solubility and dissolution improvement:
In order to overcome the limited solubility of the pharmaceuticals, these are formulated as liquisolid tablets. The method of preparation of liquisolid tablets as well as the effect of various formulation and processing variables on the preparation and the release properties of the tablets are studied by number of scientists. This technique is successfully applied for low dose water insoluble drugs. However, formulation of the high dose insoluble drugs as liquisolid tablets is one of the limitations of the liquisolid technique. In fact, when the therapeutic dose of drug is more than 50 mg, dissolution enhancement in the presence of low levels of hydrophilic carrier and coating material is not significant. But by adding some materials such as polyvinyl pyrrolidone (PVP) to liquid medication (Microsystems), it would be possible to produce dry powder formulations containing liquid with high concentration of drug. By adding such materials to the liquid medication, low amount of carrier is required to obtain dry powder with free flow ability and good compatibility.11
Flowability and compressibility:
Liquisolid compacts possess acceptable flow ability and compressibility properties. They are prepared by simple blending with selected powder excipients referred to as the carriers and the coating materials. Many grades of cellulose, starch, lactose, etc. can be used as carriers, whereas silica’s of very fine particle size can be used as coating materials.
In order to have acceptable flow ability and compatibility for liquisolid powder formulation, high levels of carrier and coating materials should be added and that in turn will increase the weight of each tablet above 1 gm which is very difficult to swallow. Therefore, in practice it is impossible with conventional method to convert high dose drugs to liquisolid tablet with the tablet weight of less than 1 gm.
In such systems, the drug existed in a molecular state of subdivision and systems were free flowing, on-adherent, dry looking powders. In further studies, compression enhancers were added to these powdered solutions like microcrystalline cellulose. However, the compression of these latter systems resulted in a significant ‘Liquid Squeezing Out’ phenomenon
In this system liquid medication is to be mixed with the excipients and then compressed to tablets. It was proved that the smaller the drug concentration in the liquid medication, the more rapid the release rates, since drugs in a high concentration tend to precipitate within the polymers pores .Polymers possessing large surface areas, and diluents like microcrystalline cellulose of fine particle size and granular grades produced good flow and compression properties, resulting in acceptable tablets.
Designing of sustained release tablet:
Development of sustained release oral dosage forms is beneficial for optimal therapy in terms of efficacy, safety and patient compliance. Ideally, a controlled release dosage form will provide therapeutic concentration of the drug in the blood that is maintained throughout the dosing interval. There are several techniques for the preparation of sustained release formulations, among which control of drug dissolution is one of the best and most successful methods due to its simplicity and low cost. To achieve this aim, several methods have been developed such as preparation of salt form of drug, coating with special materials and incorporation of drugs into hydrophobic carriers.
Liquisolid technique is a new and promising method that can change the dissolution rate of drugs. It is claimed that if hydrophobic carriers such as Eudragit RL and RS are used instead of hydrophilic carries in liquisolid systems, sustained release systems can be obtained. Therefore, it is suggested that the method have the potential to be optimized for the reduction of drug dissolution rate and thereby production of sustained release systems.
Bioavailability improvement:
In the liquisolid and powdered solution systems the drug might be in a solid dosage form, it is held within the powder substrate in solution, or in a solubilised, almost molecularly dispersed state. Therefore, due to their significantly increased wetting properties and surface of drug available for dissolution, liquisolid compacts of water- insoluble substances may be expected to display enhanced drug release properties, and consequently, improved bioavailability.
Mechanism of enhanced drug release from liquisolid systems:
The three recommended mechanisms include an increased surface area of drug available for release, an increased aqueous solubility of the drug, and an improved wettability of the drug particles.
Increased drug surface area:
When the drug within the liquisolid system is absolutely dissolved in the liquid vehicle it is positioned in the powder substrate in a solubilised, molecularly dispersed state. Therefore, the surface area of drug available for release is much greater than that of drug particles within directly compressed tablets.
Consequently, with increasing drug content beyond the solubility limit and thus, increasing fraction of undissolved drug in the liquid vehicle the release rate decreases. It has been pragmatic with various drugs that the release rates are directly proportional to the fraction of the molecularly dispersed drug (FM) in the liquid formulation. FM is defined by Spireas as the ratio between the drug's solubility (Sd) in the liquid vehicle and the actual drug concentration (Cd) in this vehicle carried by each system.
Therefore,
Sd/Cd Where FM =1 if Sd ≥ Cd
Increased aqueous solubility of the drug:
In addition to the first mechanism of drug release enhancement it is expected that Cs, the solubility of the drug, might be increased with liquisolid systems. In fact, the relatively small amount of liquid vehicle in a liquisolid compact is not sufficient to increase the overall solubility of the drug in the aqueous dissolution medium. However, at the solid/liquid interface between an individual liquisolid primary particle and the release medium it is possible that in this microenvironment the amount of liquid vehicle diffusing out of a single liquisolid particle together with the drug molecules might be sufficient to increase the aqueous solubility of the drug if the liquid vehicle acts as a co solvent. The overall increase in the solubility of drugs caused by liquisolid system.
Improved wetting properties:
Due to the fact that the liquid vehicle can either act as surface active agent or has a low surface tension, wetting of the liquisolid primary particles is improved. Wettability of these systems has been confirmed by measurement of contact angles and water rising times.
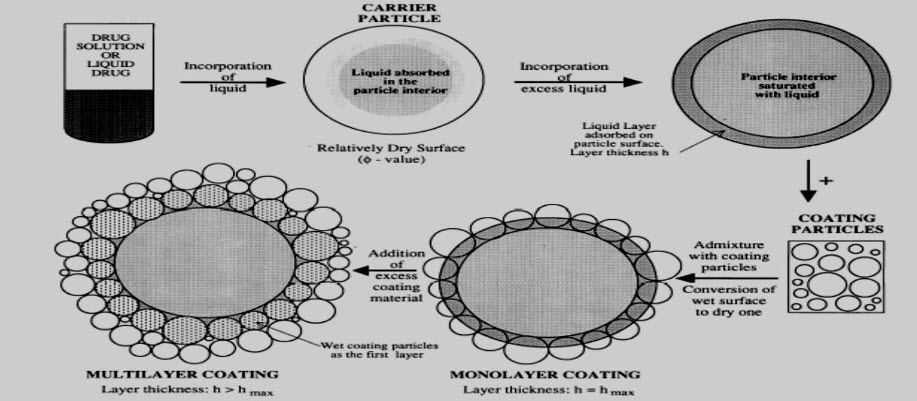
Fig: Mechanism represents formulation of liquisolid system
Theoretical aspects for designing the liquisolid systems:
The amounts of excipients (carrier and coating materials) used to prepare liquisolid compacts depends on the flowable liquid retention potential values (?-value) and the liquid loading factors (Lf), Equation (1). The ?-value of a powder is the maximum amount of a given non-volatile liquid that can be retained inside powder bulk (w/w) while maintaining acceptable flowability whereas, Lf is the mass ratio (w/w) of the liquid medication to the carrier powder in the liquisolid formulation and it is given by equation 2. Therefore, in order to calculate the required weight of excipients, we need to determine the liquid retention potential value for both carrier (?CA-value) and coating (?CO-value) materials for each non-volatile liquid vehicle used, these values are constant for the given vehicle/powder system. Knowing the carrier: coating ratio (R), liquid loading factor (Lf) can be calculated by the following equation:
Lf = ΦCA+ ΦCO (1 / R)……..… [1]
Once liquid loading factors are obtained for such non-volatile liquid vehicle used in this study, the optimum weight of the carrier (Q), required for the respective vehicle could be calculated by using the Equation (2).
Lf = W / Q……………………. [2]
Where: W is the weight of the liquid medication (the drug + non-volatile liquid vehicle) Once the values for Q are obtained for the respective vehicle, the optimum weight of the coating material (q) could also be obtained (Equation 3).
R=Q/q……………………..…. [3]
Where
Q = Amount of carrier material
q = Amount of coating material
Angle of Slide:
Required amount of carrier is weighed and placed at one end of a metal plate with a polished surface. The other end is gradually raised till the plate becomes angular to the horizontal at which powder is about to slide. This angle on which the material is about to slide from the plate is known as angle of slide. It is used as a measure of the flow properties of powders. Angle of 330 is regarded as optimum.12
Flowable liquid retention potential (Φ value):-
The Flowable Liquid Retention Potential of a powder is defined as the maximum amount of a given non-volatile liquid that can be retained inside its bulk (w/w) while maintaining acceptable flowability. It is denoted by Φ.
The Compressible Liquid Retention Potential (Ψ) of a powder is the maximum amount of liquid, the powder can retain inside its bulk (w/w) while maintaining acceptable compactability, to produce compacts of suitable hardness and friability, with no liquid squeezing out phenomenon during the compression process.
The Φ values are calculated according to equation:
Weight of liquid
Φ value = _______________
Weight of solid
Contact Angle:
The contact angle is the angle, conventionally measured through the liquid, at which a liquid/vapor interface meets a solid surface. It quantifies the wettability of a solid surface by a liquid: if the contact angle is small, a drop of the liquid will spread on the solid; if the contact angle is large, the drop of liquid will bead up.
For assessment of wettability, contact angle of Liquisolid tablets is measured. To measure the contact angle, a drop of liquid is directly placed on a flat surface of the solid in the so called imaging method. The liquid drop is prepared using a saturation solution of the drug in Simulated Gastric Fluid, Simulated Intestinal Fluid media and an excessively large amount of pure drug is added to this media, shaken for 24 hours at a constant rate then the upper solution was centrifuged. A drop of this solution was placed on the surface of the tablet. By measuring the height and diameter of the sphere drop on the liquisolid tablets and direct compressed tablets, contact angle is measured. Liquisolid tablet contact angle is less than that of direct compressed tablets. Polysorbate 80 showed the lowest contact angle in the liquisolid tablets.
CONCLUSION:
Liquisolid compact technique could be effectively used to prepare rapid release tablets of water insoluble drugs.
The liquisolid compacts were prepared using MCC and silica as carrier and coating material and PEG 400 was used as a liquid vehicle. In this technique drug is dissolved in a non volatile solvent and by this liquid medicament is converted to non adherent, dry looking and free flowing by using suitable carrier and coating material.
The flow properties of liquisolid compacts showed an acceptable flowability. The hardness, friability, weight variation and disintegration tests were within acceptable limit. The in vitro dissolution study confirmed enhanced drug release fromliquisolid compacts. The higher dissolution rate showed by Liquisolid compacts may imply enhanced oral bioavailability due to the increased wetting properties and surface of drug available for dissolution.
The solubility-dissolution behavior is the rate-limiting step to absorption from the gastrointestinal tract of poorly water soluble drugs and needs to be enhanced.
REFERENCES:
1. Sathali Hasan Abdul A., and Deepa C., Formulation of Liquisolid tablets of Candesartan Cilexetil, Int. J. of Research in Pharmaceutical Sciences (IJRPS).2013; 4:(2), 238-249.
2. Rao Sambasiva A., Naga Aparna. T., Liquisolid Technology: An Overview, Int. J. of Research in Pharmaceutical and Biomedical Sciences (IJRPBS). 2011; 2:(2), 401-409.
3. Akinlade Babatunde, Elkordy A., Amal, Essa A., Ebtessam, Elhagar Sahar., Liquisolid Systems to improve the Dissolution of Furosemide, Scientia Pharmaceutica. 2010; 78, 325-344.
4. Chaudhary Amit, Nagaich Upendra, Gulati Neha, Sharma V. K., and Khosa R. L., Enhancement of solubilization and bioavailability of poorly soluble drugs by physical and chemical modifications: A recent review, J. of Advanced Pharmacy Education & Research (JAPER). 2012; 2:(1), 32-67.
5. Khalid M. El-Say, Ahmed M. Samy, Mohamed I. Fetouh., Formulation and evaluation of Rofecoxib liquisolid tablets, Int. J. of Pharmaceutical Sci. Research and Review (IJPSRR). 2010; 3:(1), 135-142.
6. Spireas S., Liquisolid systems and methods of preparing the same, Pharmaceutical Patent. 2002.
7. Javadzadeh Y., jafri B-navimipour and nokhodchi A., Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine), Int. J.Pharm. 2007; 341 :(1-2), 26-34.
8. Kulkarni Ajit S., Aloorkar Nagesh H., Mane Madhav S., and Gaja Jayashree B., Liquisolid systems: A Review, Int. J. of Pharmaceutical sciences and Nanotechnology (IJPSN). 2010; 3:(1), 795-802.
9. Kasture SV., Gondkar SB., Darekar AB., Dash P. and Bhambar KV., Enhancement of dissolution rate of Lansoprasole using liquisolid tablet technique, Int. J. of pharmaceutical research (IJPR). 2011; 3:(1), 27-31.
10. Rajesh K., Rajalakshmi R., Umamaheswari J., Kumar Ashok C.K., Liquisolid Technique a novel approach to enhance solubility and bioavailability, Int. J. of Biopharmaceutics (IJB). 2011; 2:(1), 8-13.
11. Yadav V.B., Yadav A.V., liquisolid granulation technique for tablet manufacturing: An overview, J. of pharmacy research (JPR). 2009; 2:(4), 670-674.
12. Tayel SA., Soliman I., and Louis D., Improvement of dissolution properties of carbamazepine through application of the liquisolid tablet technique, Eur J Pharm Biopharm. 2008; 69, 342-347.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








