
 About Authors:
About Authors:
V. Ravi Chandra
Research Associate
Parexel
B.Pharmacy from Priyadarshini College of pharmacy,
Osmania University,
Hyderabad, AP, INDIA
Abstract:
Huntington's disease is an intricate illness. It is a neurodegenerative, insidious disorder-which means it is harmful and fatal but gradually. The onset of the disease is very late to diagnose the disease. Huntington's disease (HD) also called as ‘Huntington’s Chorea’. Chorea means dance in Greek, which is aftereffect of genetically programmed degeneration of brain cells, called neurons, in certain areas of the brain. This degeneration causes uncontrolled movements, loss of intellectual faculties, and emotional disturbance. By choreic movement physically we can diagnose HD. HD is also a familial disease, passed from parent to child through a mutation in the normal gene. Attempts to study early disease are not unique in neurology (e.g., mild cognitive impairment and vascular cognitive impairment), but studying otherwise healthy-appearing individuals who have nearly 99% certainty of manifesting the symptoms of brain disease does provide distinct but valuable information about the true natural history of the disease. A genetic test, coupled with a complete medical history and neurological and laboratory tests, helps physicians diagnose HD. Presymptomic testing is available for individuals who are at risk for carrying the HD gene. HD occurs in about one out of every 10,000 Caucasian individuals. In India, the cases are unknown because of failure of doctors to diagnose HD as they never seen HD in their practice. Approximately 2.5 times more individuals are at risk for the disorder because of the midlife peak in age at onset.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1222
Introduction
Huntington’s disease is an autosomal-dominant, progressive neurodegenerative disorder with a distinct phenotype, including chorea and dystonia, in coordination, cognitive decline, and behavioral difficulties. Typically, onset of symptoms is in middle age after affected individuals have had children, but the disorder can manifest at any time between infancy and senescence. The mutant protein in Huntington’s disease—huntingtin—results from and expanded CAG repeat leading to a polyglutamine strand of variable length at the N-terminus. Evidence suggests that this tail confers a toxic gain of function. The precise pathophysiological mechanisms of Huntington’s disease are poorly understood, but research in transgenic animal models of the disorder is providing insight into causative factors and potential treatments. Several doctors, but George Huntington’s vivid description led to the eponymous designation of the disorder as Huntington’s disease. Over the next, few decades noted the hereditary nature of chorea in the 19thcentury, the worldwide distribution of the disorder and its juvenile form were recorded. The discovery of the causal HDgene has stimulated research, and work is now focusing on molecular mechanisms of disease.
Historical introduction
Huntington’s disease was given different names throughout this history as understanding of the disease changed. Originally called simply 'chorea' for the jerky dance-like movements associated with the disease, HD has also been called 'hereditary chorea' and 'chronic progressive chorea'.
The first thorough description of the disease was by George Huntington in 1872. Examining the combined medical history of several generations of a family exhibiting similar symptoms, he realized their conditions must be linked; he presented his detailed and accurate definition of the disease as his first paper.

Pathophysiology
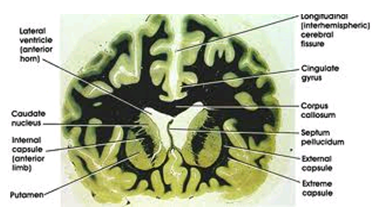
The most striking pathology in HD occurs within the neostriatum, in which gross atrophy of the caudate nucleus and putamen is accompanied by selective neuronal loss and astrogliosis. Marked neuronal loss also is seen in deep layers of the cerebral cortex. Other regions, including the globus pallidus, thalamus, sub thalamic nucleus, substantia nigra, and cerebellum, show varying degrees of atrophy depending on the pathologic grade. The extent of gross striatal pathology, neuronal loss, and gliosis provides a basis for grading the severity of HD pathology. No gross striatal atrophy is observed in grades 0 and 1. Grade 0 cases have no detectable histologic neuropathology in the presence of a typical clinical picture and positive family history suggesting HD. Grade 1 cases have neuropathologic changes that can be detected microscopically but without gross atrophy. In grade 2, striatal atrophy is present, but the caudate nucleus remains convex. In grade 3, striatal atrophy is more severe, and the caudate nucleus is flat. In grade 4, striatal atrophy is most severe, and the medial surface of the caudate nucleus is concave.
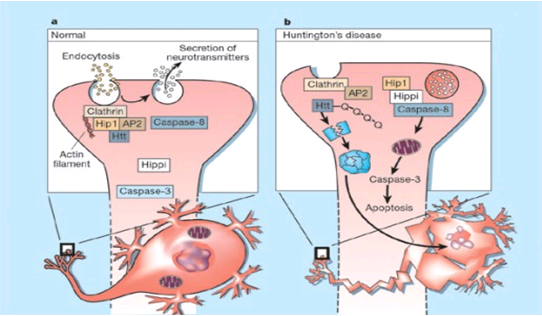
The genetic basis of HD is the expansion of a cysteine-adenosine-guanine (CAG) repeat encoding a polyglutamine tract in the N -terminus of the protein product called huntingtin. The function of huntingtin is not known. Normally, it is located in the cytoplasm. The association of huntingtin with the cytoplasmic surface of a variety of organelles, including transport vesicles, synaptic vesicles, microtubules, and mitochondria, raises the possibility of the occurrence of normal cellular interactions that might be relevant to neuro-degeneration. N-terminal fragments of mutant huntingtin accumulate and form inclusions in the cell nucleus in the brains of patients with HD, as well as in various animal and cell models of HD. The presence of neuronal intranuclear inclusions (NIIs) initially led to the view that they are toxic and, hence, pathogenic.More recent data from striatal neuronal cultures transfected with mutant huntingtin and transgenic mice carrying the spinocerebellar ataxia-1 (SCA-1) gene (another CAG repeat disorder) suggest that NIIs may not be necessary or sufficient to cause neuronal cell death, but translocation into the nucleus is sufficient to cause neuronal cell death.Caspase inhibition in clonal striatal cells showed no correlation between the reduction of aggregates in the cells and increased survival.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Epidemiology
HD affects males and females in relatively equal numbers. The disorder occurs in various geographic and ethnic populations worldwide. The frequency of HD appears to vary among different populations, ranging from an estimated 4 to 10 individuals in 10,000.
Huntington’s disease shows a stable prevalence in most populations of white people of about 5–7 affected individuals per 100 000. Exceptions can be seen in areas where the population can be traced back to a few founders, such as Tasmania and the area around Lake Maracaibo in Venezuela. In Japan, prevalence of the disorder is 0.5 per 100 000, about 10% of that recorded elsewhere, and the rate is much lower in most of Asia. African populations show a similarly reduced prevalence, although in areas where much intermarriage with white people takes place the frequency is higher. Currently, the higher incidence of Huntington’s disease in white populations compared with African or Asian people relates to the higher frequency of huntingtin alleles with 28–35 CAG repeats in white individuals.
In people with dentatorubropallidoluysian atrophy, which is frequent in Asia, expanded alleles for the causal gene (ATN1) are much more typical in Asian populations. Why do population differences in huntingtin alleles persist? What is the genetic fitness of Huntington’s disease? Findings have shown no consistent increase or decrease in the number of children of affected individuals. Furthermore, the HDgene does not seem to confer any promising health benefits other than a possible lower incidence of cancer, perhaps related to an up regulation of TP53in Huntington’s disease. No data suggest that expanded huntingtin alleles protect against epidemic infectious disease. The late onset of Huntington's disease means it does not usually affect reproduction. The prevalence is similar for men and women, but varies greatly geographically because of ethnicity, local migration and past immigration patterns. The rate of occurrence is highest in peoples of Western European descent, averaging around seventy per million people, and is lower in the rest of the world, e.g. one per million people of Asian and African descent. Additionally, some localized areas have a much higher prevalence than their regional average.
Frequency
Estimates of the prevalence of HD in the United States range from 4.1-8.4 per 100,000 people. Accurate estimates of the incidence of HD are not available. The frequency of HD in different countries varies greatly. A few isolated populations of western European origin have an unusually high prevalence of HD that appears to have resulted from a founder effect. These include the Lake Maracaibo region in Venezuela (700 per 100,000 people), the island of Mauritius off the South African coast (46 per 100,000 people), and Tasmania (17.4 per 100,000 people). The prevalence in most European countries ranges from 1.63-9.95 per 100,000 people. The prevalence of HD in Finland and Japan is less than 1 per 100,000 people.
Symptoms
Huntington’s disease is a widely variable disorder, even within the same family. The early symptoms of Huntington’s disease generally include:
• Slight, uncontrollable muscular movements
• Chorea
• stumbling and clumsiness
• lack of concentration
• lapses of short-term memory
• depression
• changes of mood, sometimes including aggressive or antisocial behavior
The rate of progress of Huntington’s disease varies, but generally, it develops over 15-25 years.
Later in the illness, people may experience different symptoms, which include:
• Involuntary movements
• Difficulty in speech and swallowing
• Weight loss as well as emotional changes, resulting in:
• Stubbornness
• Frustration
• Mood swings
• Depression
Chorea (derived from the Greek word meaning to dance) is the most common movement disorder seen in HD. Chorea is a characteristic feature of HD and, until recently; the disorder commonly was called Huntington chorea. Chorea, as defined by the World Federation of Neurology, is a state of excessive, spontaneous movements, irregularly timed, randomly distributed, and abrupt. Severity of chorea may vary from restlessness with mild intermittent exaggeration of gesture and expression, fidgeting movements of the hands, and unstable dance like gait to a continuous flow of disabling violent movements. Initially, mild chorea may pass for fidgetiness. Severe chorea may appear as uncontrollable flailing of the extremities (i.e., ballism), which interferes with function.

Cognitive decline is characteristic of HD, but the rate of progression among individual patients can vary considerably. Dementia and the psychiatric features of HD are perhaps the earliest and most important indicators of functional impairment. The dementia syndrome associated with HD includes early onset behavioral changes, such as irritability, untidiness, and loss of interest.
Slowing of cognition, impairment of intellectual function, and memory disturbances are seen later. This pattern corresponds well to the syndrome of sub cortical dementia, and it has been suggested to reflect dysfunction of frontal-sub cortical neuronal circuitry. Early stages of HD are characterized by deficits in short-term memory, followed by motor dysfunction and a variety of cognitive changes in the intermediate stages of dementia. These deficits include diminished verbal fluency, problems with attention, executive function, visuospatial processing, and abstract reasoning.
Language skills become affected in the final stages of the illness, resulting in a marked word-retrieval deficit. The behavioral disorder of HD is represented most commonly by affective illness. Depression is more prevalent, with a small percentage of patients experiencing episodic bouts of mania characteristic of bipolar disorder. Patients with HD and persons at risk for HD may have an increased rate of suicide. Patients with HD also can develop psychosis, obsessive-compulsive symptoms, sexual and sleep disorders and changes in personality.
Causes
The selective neuronal dysfunction and subsequent loss of neurons in the striatum, cerebral cortex, and other parts of the brain can explain the clinical picture seen in cases of HD. Several mechanisms of neuronal cell death have been proposed for HD, including excitotoxicity, oxidative stress, impaired energy metabolism, and apoptosis.
Excitotoxicity
Excitotoxicity refers to the neurotoxin effect of excitatory amino acids in the presence of excessive activation of postsynaptic receptors. Intrastriatal injections of kainic acid, an agonist of a subtype of glutamate receptor, produce lesions similar to those seen in HD.
Intrastriatal injections of quinolinic acid, an N-methyl-D-aspartate (NMDA) receptor agonist, selectively affect medium-sized GABA-ergic spiny projection neurons, sparing the striatal interneuron’s and closely mimicking the neuropathology seen in HD. NMDA receptors are depleted in the striata of patients with HD, suggesting a role of NMDA receptor-mediated excitotoxicity, but no correlation exists between the distribution of neuronal loss and the density of such receptors. The theory that reduced uptake of glutamate by glial cells may play a role in the pathogenesis of HD also has been proposed.
Oxidative stress
Oxidative stress is caused by the presence of free radicals (i.e., highly reactive oxygen derivatives) in large amounts. This may occur as a consequence of mitochondrial malfunction or excitotoxicity and can trigger apoptosis. Striatal damage induced by quinolinic acid can be ameliorated by the administration of spin-trap agents, which reduce oxidative stress, providing indirect evidence for the involvement of free radicals in excitotoxic cell death.
Impaired energy metabolism
Impaired energy metabolism reduces the threshold for glutamate toxicity and can lead to activation of excitotoxic mechanisms as well as increased production of reactive oxygen species. Nuclear magnetic resonance spectroscopy studies have shown elevated lactate levels in the basal ganglia and occipital cortex of patients with HD. Patients with HD have an elevated lactate-pyruvate ratio in the cerebrospinal fluid. A reduction in the activity of the respiratory chain complex II and III (and less in complex IV) of mitochondria of caudate neurons in patients with HD has been reported.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Clinical findings
Individuals with Huntington’s disease can become symptomatic at any time between the ages of 1 and 80 years; before then, they are healthy and have no detectable clinical abnormalities. This healthy period merges imperceptibly with a prediagnostic phase, when patients show subtle changes of personality, cognition, and motor control. Both the healthy and prediagnostic stages are sometimes called presymptomatic, but in fact, the prediagnostic phase is associated with findings, even though patients can be unaware of them. Diagnosis takes place when findings become sufficiently developed and specific. In the prediagnostic phase, individuals might become irritable or disinherited and unreliable at work; multitasking becomes difficult and forgetfulness in addition, anxiety mounts. Family members note restlessness or fidgeting, sometimes keeping their partners awake at night. Eventually, this stage merges with the diagnostic phase, during which time affected individuals show distinct chorea, in coordination, motor impersistence, and slowed saccadic eye movements.
Cognitive dysfunction in Huntington’s disease, often spares long-term memory but impairs executive functions, such as organizing, planning, checking, or adapting alternatives, and delays the acquisition of new motor skills. These features worsen over time; speech deteriorates faster than comprehension. Unlike cognition, psychiatric and behavioral symptoms arise with some frequency but do not show stepwise progression with disease severity. Depression is typical and suicide is estimated to be about five to ten times that of the general population (about 5–10%). Manic and psychotic symptoms can develop. Suicidal ideation is a frequent finding in patients with Huntington’s disease. In a cross-sectional study, about 9% of asymptomatic at-risk individuals contemplated suicide at least occasionally, perhaps a result of being raised by an affected parent and awareness of the disease. In the prediagnostic phase, the proportion rose to 22%, but in patients who had been recently diagnosed, suicidal ideation was lower. The frequency increased again in later stages of the illness.
The correlation of suicidal ideation with suicide has not been studied in people with Huntington’s disease, but suicide attempts are not uncommon. In one study, researchers estimated that more than 25% of patients attempt suicide at some point in their illness. Individuals without children might be at amplified risk, and for these people access to suicidal means (i.e., drugs or weapons) should be restricted. The presence of affective symptoms, specific suicidal plans, or actions that increase isolation (e.g., divorce, giving away pets) warrants similar precautions. Although useful for diagnosis, chorea is a poor marker of disease severity. Patients with early onset Huntington’s disease might not develop chorea, or it might arise only transiently during their illness. Most individuals have chorea that initially progresses but then, with later onset of dystonia and rigidity, it becomes less prominent.
Inheritance
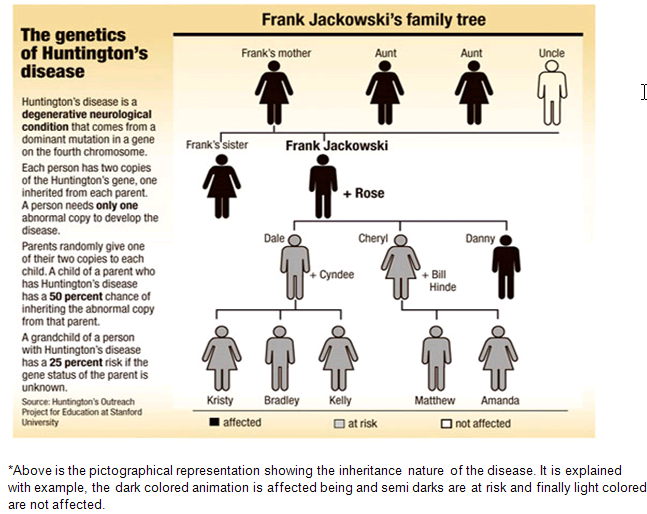
Huntingtin
The use of Huntington's disease is still not fully understood. What is known about Huntington's disease is that it occurs due to mutations in a single gene coding for a protein called huntingtin.
Huntingtin is expressed in all human and mammalian cells, with the highest concentrations in the brain and testes; moderate amounts are present in the liver, heart, and lungs. Recognizable orthologos of protein is present in many species, including zebra fish, drosophila, and slime moulds. The role of the wild-type protein is, yet, poorly understood, as is the underlying pathogenesis of Huntington’s disease. One mechanism by which an autosomal-dominant disorder such as Huntington’s disease could cause illnesses by haploin sufficiency, in which the genetic defect leads to inadequate production of a protein need for vital cell function. This idea seems unlikely because terminal deletion or physical disruption of the HDgene in man does not cause Huntington’ disease. Furthermore, one copy of the HDgene does not cause a disease phenotype in mice. Whereas homozygous absence of the HDgene is associated with embryonic lethality in animals, people homozygous for the HDgene have typical development. Findings suggest that the mutant HDgene confers toxic gain of function. A persuasive line of evidence for this idea comes from nine other known human genetic disorders with expanded (and expressed) polyglutaminerepeats: spinocerebellar ataxia types, Dentatorubropallidoluysian atrophy, and spinobulbar muscular atrophy. For none of these disorders is there evidence to suggest an important role for haplo insufficiency.
In spinobulbar muscular atrophy, complete deletion of the androgen receptor is not associated with neuromuscular disease. All nine diseases show neuronal inclusions containing aggregates of polyglutamines and all have a pattern of selective neurodegeneration. One of the most striking features of these disorders is the robust inverse correlation between age of onset and number of polyglutamine repeats. Results suggest that the length of the polyglutamine repeat indicates disease severity irrespective of the gene affected, with the longest repeat lengths associated with the most disabling early-onset (juvenile) forms of these disorders. Although difficult to confirm, some data also suggest that the rate of progression might be faster with longer CAG repeats, particularly for individuals with juvenile-onset disease. The most convincing evidence for a gain of function in Huntington’s disease is the structural biology of polyglutamine strands. In-vitro evidence suggests that Polyglutamines will begin to aggregate, initially by forming dimmers, trimmers, and oligomers.
This process needs a specific concentration of protein and a minimum of 37 consecutive glutamine residues, follows a period of variable abeyance and proceeds faster with higher numbers of glutamine repeats. These findings might account for both delayed onset of disease and the close correlation with polyglutamine length. The rate of aggregation increases with the number of glutamine residues, which accords with evidence showing that length of expansion is associated with early age of onset.
Juvenile HD
Juvenile HD (Westphal variant), defined as having an age of onset of younger than 20 years, is characterized by parkinsonian features, dystonia, long-tract signs, dementia, epilepsy, and mild or even absent chorea. Juvenile HD is characterized by a movement disorder, which differs from that of affected adults. The earlier the disease onset, the more likely it is that the child will be very rigid, and the less likely it is that he or she will have the chorea (involuntary movements) seen in most adults.
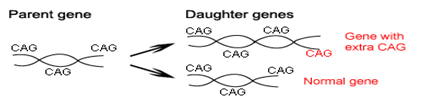
In 25-30% of cases, there is a tendency to epileptic seizures —something almost never seen in adults with HD. Juvenile HD also involves changes in behavior and mental function. Like its adult counterpart, juvenile HD remains incurable, and there are no treatments, which can stop or slow the course of the disease. However, dramatic advances in research have given rise to tremendous optimism that new forms of therapy will soon be within sight. Huntington disease is caused by a genetic mutation called a “CAG repeat expansion” — this type of mutation was unknown until the 1990s. The letters CAG refer to three of the four nucleotides which make up DNA — A (adenine), T (thymine), (guanine), C (cytosine) —and HD is one of a group of neurological diseases caused by this type of gene mutation. In HD, there is an expanded CAG repeat in a gene on chromosome. The function of this gene remains unknown. People who do not have HD have approximately 10-35 CAG repeats in this particular gene, while persons with HD usually have more than 40 repeats.
A very small number of individuals with 35-40 repeats may or may not develop symptoms, or may live well into their 60s or 70s without developing symptoms of the disease. Juvenile HD involves a larger number of repeats — 50, 60 or more— than is found in the adult-onset form of the disease. Generally, the more repeats, the earlier the disease tends to start, but it is not possible to predict the exact age of onset for any individual on the basis of the number of CAG repeats. AG repeat length can change as a parent passes the abnormal HD gene to a child. Thus, a parent whose HD gene has 45 repeats can have a child with 44, 48, or 52 repeats. The number of repeats tends to get bigger, although occasionally it can get smaller. Very large increases in CAG repeat length usually come from an affected father, and juvenile HD is more likely to be seen in the children of HD fathers than HD mothers. There may be other mutations in the HD gene, which do not involve the CAG repeat, and there may be “modifier” genes, which interact with the HD gene to influence the onset and course of the disease.
Diagnosing juvenile Huntington disease can be extremely difficult. Juvenile HD is extremely rare, and few physicians will have encountered the disease before. This can lead to a great deal of time being spent eliminating other possibilities. Compounding this problem, of course, the examination of children can itself be challenging, and if the child is frightened and will not walk or talk for the doctor, signs of early impairment may not be evident. The best physician may have to see the child several times before being confident that neurological symptoms are apparent. A neurologist can usually determine that the child has a disorder affecting a portion of the brain called the basal ganglia and may suspect that it is progressive, but it can be difficult to distinguish HD from other diseases.
A complete and accurate family history is invaluable in evaluating a child with symptoms suggestive of Huntington disease, and can make the process of diagnosis much more straightforward. However, there are situations in which parents may not even be aware that HD is in the family; in other cases, an adopted child may be involved. A number of tests may be used in conjunction with the presence or absence of a family history, and clinical presentation, to help clarify the diagnosis.
Diagnosis
Diagnosis of HD is not an easy work. This requires an extensive care while diagnosing. The discovery of the HD gene in 1993 resulted in a direct genetic test to make or confirm a diagnosis of HD in an individual who is exhibiting HD-like symptoms. Using a blood sample, the genetic test analyzes DNA for the HD mutation by counting the number of repeats in the HD gene region. Individuals who do not have HD usually have 28 or fewer CAG repeats. Individuals with HD usually have 40 or more repeats. A small percentage of individuals, however, have a number of repeats that fall within a borderline region.
A neurologist will interview the individual intensively to obtain the medical history and rule out other conditions. A tool used by physicians to diagnose HD is to take the family history, sometimes called a pedigree or genealogy. It is extremely important for family members to be candid and truthful with a doctor who is taking a family history. The doctor will also ask about recent intellectual or emotional problems, which may be indications of HD, will test the person's hearing, eye movements, strength, coordination, involuntary movements (chorea), sensation, reflexes, balance, movement, and mental status, and will probably order a number of laboratory tests as well. People with HD commonly have impairments in the way the eye follows or fixes on a moving target.
Abnormalities of eye movements vary from person to person and differ, depending on the stage and duration of the illness. The discovery of the HD gene in 1993 resulted in a direct genetic test to make or confirm a diagnosis of HD in an individual who is exhibiting HD-like symptoms. Using a blood sample, the genetic test analyzes DNA for the HD mutation by counting the number of repeats in the HD gene region. Individuals who do not have HD usually have 28 or fewer CAG repeats. Individuals with HD usually have 40 or more repeats. A small percentage of individuals, however, have a number of repeats that fall within a borderline region.
Doctors/physicians would use medical imaging methods for diagnosis of the disease. The various types of diagnostic methods are as follows:
· Computerized Tomography (CT)
· Magnetic Resonance Imaging (MRI)
· Positron Emission Tomography (PET)
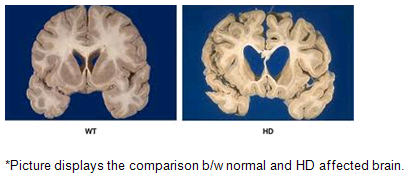
Above-mentioned imaging techniques help to understand the brain anatomically well. HD affected brain differs with the normal brain as shown in the below picture.
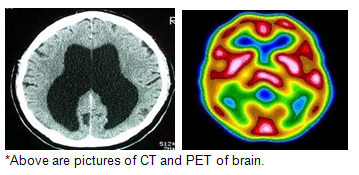
PET visualizes metabolic or chemical abnormalities in the body, and investigators hope to ascertain if PET scans can reveal any abnormalities that signal HD. Investigators conducting HD research are also using PET to characterize neurons that have died and chemicals that are depleted in parts of the brain affected by HD.
Like PET, a form of magnetic resonance imaging (MRI) called functional MRI can measure increases or decreases in certain brain chemicals thought to play a key role in HD. Functional MRI studies are also helping investigators understand how HD kills neurons in different regions of the brain.
Imaging technologies allow investigators to view changes in the volume and structures of the brain and to pinpoint when these changes occur in HD. Scientists know that in brains affected by HD, the basal ganglia, cortex, and ventricles all show atrophy or other alterations.
Treatment
There is no cure for Huntington's disease, and there is no known way to stop the disease from getting worse. The goal of treatment is to slow down the symptoms and help the person function for as long and as comfortably as possible.
Medications vary depending on the symptoms. Most common medications as follows:
Antipsychotic(hallucinations, delusions, violent outbursts): haloperidol, chlorpromazine, olanzapine (contraindicated if patient has dystopia)
Antidepressants(depression, obsessive-compulsive behavior): fluoxetine, sertraline hydrochloride, nortriptyline
Tranquilizers (anxiety, chorea): benzodiazepines, paroxetine, venlafaxin, beta-blockers
Mood-stabilizers(mania, bipolar disorder): lithium, valproate, carbamazepine
In August 2008, the U.S. Food and Drug Administration approved tetrabenazine to treat chorea, making it the first drug approved for use in the United States to treat the disease.
Tests:
Presymptomatic testing: Presymptomatic testing is used for people who have a family history of HD but have no symptoms themselves. If either parent had HD, the person's chance would be 50-50. In the past, no laboratory test could positively identify people carrying the HD gene--or those fated to develop HD--before the onset of symptoms.
Usually includes sessions devoted to genetic counseling, a neurological exam, a psychological interview, discussion of the results, and follow-up. Neurological exam is meant to determine whether the patient has any symptoms, in which case they may choose to discontinue testing procedure. Sessions are meant to ensure that the person about to undergo testing understands the implications of the knowledge of the results.
The testing process is a simple blood test. The blood the presence or absence of the HD mutation. It is encouraged that patients have either a blood sample from a family member who has HD or the results of his/her genetic test for confirming the diagnosis.
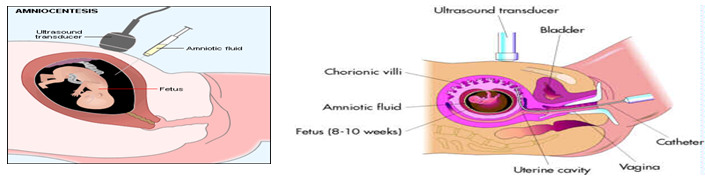
Prenatal testing: Prenatal testing is one of a range of options, which may be of interest to couples who are at risk of passing the disease-causing version of the HD gene to a child. Amniocentesis involves testing a sample of amniotic fluid from the womb. Usually done when woman is between 16 and 20 weeks during pregnancy.
Chorionic Villi Sampling: Performed earlier than amniocentesis - between the 10th and 12th weeks of pregnancy. In CVS, a catheter or thin needle is inserted into the womb to extract some of the chorionic villi - cells from the tissue that will become the placenta. The chorionic villi contain the same chromosomes as the fetus.
Conclusion
The basic conclusion is awareness and spreading awareness, as there is no panacea yet. In 1968, after experiencing HD in his wife's family, Dr. Milton Wexler was inspired to start the Hereditary Disease Foundation (HDF), with the aim of curing genetic illnesses by coordinating and supporting research. The foundation and Dr. Wexler's daughter, Nancy Wexler, were key parts of the research team in Venezuela, which discovered the HD gene. As of 2009, Nancy Wexler is the foundation's president.At roughly the same time as the HDF formed, Marjorie Guthrie helped to found the Committee to Combat Huntington's Disease (now the Huntington's disease Society of America), after her husband, Woody Guthrie died from complications of HD. Since then, support and research organizations have formed in many countries around the world and have helped to increase public awareness of HD. A number of these collaborate in umbrella organizations, like the International Huntington Association and the Euro HD network. Many support organizations hold an annual HD awareness event, some of which have been endorsed by their respective governments. For example, June 6 is designated "National Huntington's Disease Awareness Day" by the US senate.
References
1. Martin JB, Gusella JF: Huntington's disease-pathogenesis and management. N English J Med 1986; 315: 1267-1275.
2. Pani C, Rajadhyaksha SB, Varudkar BL et al: Juvenile Huntington's disease. Neurol India 1993; 41: 224-226.
3. Collewijn H, Went L, Tamminga ED: Coulometer defects in patients with Huntington's disease and their off springs. J Neurol Science 1988; 86: 307-320.
4. Sharma P, Savy L, Britton J: Huntington's disease a molecular genetics and CT Comparison. J Neurol Neurosurgeon Psychiatry 1996; 60: 206-208.
5. Huntington, G. (1872-04-13). “on Chorea" Medical and Surgical Reporter of Philadelphia 26 (15): 317–321.
6. “Huntington's Disease Society of America” www.hdsa.org/about/hdsa-history. Retrieved 2009-03-17
7. Walker FO (2007). "Huntington's disease". Lancet 369 (9557): 219. Doi: 10.1016/S0140-6736(07)60111-1 PMID 17240289.
8. Stober T, Wussow W, Schimrigk K. Bicaudate diameter--the most specific and simple CT parameter in the diagnosis of Huntington's disease. Neuroradiology. 1984; 26(1):25-8. (Medline)
9. Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes Cell. 1993; 72:971–983. (Pub Med).
10. Story C. Landis, director of NINDS
11. Huntington's chorea: a centenary review, KENNETH W. G. HEATHFIELD, Postgraduate Medical Journal (January 1973) 49, 32-45.
12. Harper PS: Huntington’s disease. London: Saunders, 1996, European Journal of Human Genetics (2002) 10, 689 – 693
13. Huntington G. On chorea. Med Surg Report. 1872; 26:320.
14. Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neural. May 1998; 57(5):369-84. [Medline].
15. Fahy M, C Robbins, M Bloch, et al. Different options for prenatal testing for Huntington's disease using DNA probes. J. Med Genet. 1989; 26:353-357.
16. Quarrell OWJ, AL Meredith, A Tyler, et al. Exclusion testing for Huntington's disease in pregnancy with a closely linked DNA marker. Lancet 1987; 1:1281-3.
17. Millan FA, A Curtis, M Mennie, et al. Prenatal exclusion testing for Huntington's disease: a problem of too much information. J. Med. Genet. 1989; 26:83-5.
18. Meissen GJ, RH Myers, CA Mastromauro, et al. Predictive testing for Huntington's disease with use of a linked DNA marker. N. Engl. J. Med. 1988; 318:535-42.
19. Brandt J, KA Quaid, SE Folstein, et al. Presymptomatic diagnosis of delayed-onset disease with linked DNA markers. JAMA 1989; 261:3108-14.
20. Quaid KA, J Brandt, RR Faden, SE Folstein. Knowledge, attitude and the decision to be tested for Huntington's disease. Clin. Genet. 1989; 36:431-438.
21. Morris MJ, A Tyler, L Lazarou, et al. Problems in genetic prediction for Huntington's disease. Lancet 1989; 2:601-3.
22. Craufurd D, A Dodge, L Kerzin-Storrar, et al. Uptake of Presymptomatic predictive testing for Huntington's disease. Lancet 1989; 2:603-5.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








