
 About Author:
About Author:
Kambham Venkateswarlu
Graduate Student
Sri Lakshmi Narasimha College of Pharmacy,
Palluru, Chittoor District, Andhra Pradesh-517132, India.
k.v.reddy9441701016@gmail.com
ABSTRACT:
This is an inherited arrhythmia that causes the bottom chambers of the heart (the ventricles) to beat so fast that they can prevent the blood from circulating efficiently in the body. When this situation occurs (called ventricular fibrillation), the individual will faint and may die in a few minutes if the heart is not reset. While this is a disease that usually affects people in their 30's, it has actually been described at all ages. So it is important to screen everybody in a family. Not everybody who has the disease will have arrhythmias. However, we cannot know yet who will be OK and who will have problems. If you have had fainting spells related to Brugada syndrome, our experience indicates that you are at very high risk of having them again.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1602
INTRODUCTION:
The Brugada syndrome is a genetic disease that is characterised by abnormal electrocardiogram (ECG) findings and an increased risk of sudden cardiac death. It is named by the Spanish/Belgian cardiologists Pedro Brugada and Josep Brugada. It is the major cause of sudden unexplained death syndrome (SUDS), and is the most common cause of sudden death in young men without known underlying cardiac disease in Thailand and Laos.
Although the ECG findings of Brugada syndrome were first reported among survivors of cardiac arrest in 1989, it was only in 1992 that the Brugada brothers recognized it as a distinct clinical entity, causing sudden death by causing ventricular fibrillation (a lethal arrhythmia) in the heart.
The first patient with this syndrome was seen in 1986. The patient was a three year old boy from Poland. He had presented multiple episodes of loss of consciousness and had been resuscitated multiple times by his father. The child's sister had died suddenly at age two after multiple episodes of aborted sudden death. When she died, she was receiving amiodarone and had a ventricular pacemaker implanted. The electrocardiograms of the two siblings were very similar and abnormal (picture 3). The identification of two additional patients resulted in the presentation of the preliminary data at the meeting of the North American Society of Pacing and Electrophysiology (NASPE) in 1991. The first paper including 8 patients was published in 1992. Since then, there has been an exponential increase in the number of patients recognized all over the world. The recent discovery of the genetic abnormalities linked to this syndrome, points to it being a primary electrical disease, providing an important first step in the prevention and effective treatment of this form of sudden death in patients with a structurally normal heart.
Several authors have previously reported electrocardiograms similar to the one presented in For the most part, these were considered variants of the normal electrocardiogram and no definitive link to sudden death was established.
[adsense:468x15:2204050025]
In the 1980's the CDC (Center for Disease Control) in Atlanta, reported an abnormally high incidence of sudden death in young immigrants from Southeast Asia. Interestingly, the natives knew the problem for many decades. In the Northeast of Thailand, this form of death was known as Lai Tai (death during sleep). The indigenous believe that the young men died during sleep because widow ghosts came to take them away. Many young men actually dress still as women to go to sleep at night - a practice carried out for more than 70 years - with the hope that it would mislead the widow ghost, who then would not take these young men. In Philippines the phenomenon was known as Bangungut (scream followed by sudden death during sleep) and in Japan as Pokkuri (unexpected sudden death at night). The incidence of this form of sudden death has been estimated between 26 and 38 cases per 100,000 inhabitants per year. In Laos it may cause 1 sudden death per 1.000 inhabitants per year. Unexpected sudden death is the most common cause of natural death in young Thai people. It has been only recently discovered that many of these patients suffer the Brugada syndrome. The higher prevalence of this syndrome in some areas can be explained by its genetic transmission.
ETIOLOGY AND GENETICS:
This syndrome is genetically determined. Approximately 60% of patients with (aborted) sudden death with the typical electrocardiogram have a family history of sudden death, or have family members with the same electrocardiographic abnormalities. There are also sporadic cases who are probably the patients with a de novo mutation in the family. The pattern of transmission is autosomic dominant. There is a predominance of affected males. In Thailand the disease almost exclusively affects males. The explanation to this predominance is not clear but it is possibly related to some modifier genes. Several mutations linked to this syndrome affecting the gene SCN5A which encodes for the cardiac sodium channel have been described. Some of the families studied do not present these mutations in this gene, indicating that other genetic defects will be found and that this is a genetically heterogeneous disease.
The mutations affecting patients in Japan and South Asia are still unknown. The in vitro studies show that the mutant channel has altered function, with a loss of sodium current that is temperature dependent. The loss of current leaves Ito unopposed, creating heterogeneity of refractory periods and a perfect substrate for arrhythmias based on phase 2 re-entry.
INCIDENCE:
Because the syndrome has been identified only recently, it is difficult to ascertain its incidence and distribution in the world. When we analyze the data from the different publications, the disease is responsible for 4 to 12 % of unexpected sudden deaths, and for up to 50% of all sudden death in patients with an apparently normal heart. The incidence may even be higher in the younger population. Indeed, this syndrome is the most common cause of sudden death in individuals younger than 50 in South Asia with no underlying cardiac disease. It is important to stress that the physician plays an important role in the identification of the syndrome to estimate its real prevalence. This syndrome is possibly more prevalent, but the magnitude of it has yet to be determined. The difficulty to estimate the incidence and prevalence of the disease becomes more complicated as we unravel the syndrome and some of its peculiar characteristics. It is a syndrome that can present with a typical electrocardiogram but in some other cases the patient can present concealed forms or intermittent forms, meaning that the electrocardiogram is normal at certain times. The electrocardiogram of the syndrome may be unmasked by the administration of ajmaline, flecainide or procainamide. The pharmacologic test is so specific, that it is recommended in all patients who present with a history of syncope of unknown origin or ventricular fibrillation of unknown cause. Unfortunately, these tests are not applied routinely. Without a doubt this is one of the reasons why the prevalence of the disease probably is underestimated. Recent data from France and Japan show a prevalence of 1 per 1000 electrocardiograms compatible with the syndrome in the normal adult population.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
This syndrome has already been recognized in virtually all parts of the world. We suspect that the lack of cases in some countries is due more to the lack of recognition than to the absence of the disease. With present data, the disease seems spread all over the world, a not surprising finding, given the high mobility of the population and the genetic basis of the disease. In the future we only can expect a sizeable increase in the number of identified cases as the recognition of the disease grows.
A prospective study of an adult Japanese population (22.027 subjects) showed an incidence of 0.05% of electrocardiograms compatible with the syndrome (12 subjects). A second study of adults in Awa (Japan) showed an incidence of 0.6 % (66 cases out of 10.420). However, a third study in children from Japan showed an incidence of electrocardiograms compatible with the syndrome of only 0.0006% (1 case in 163.110). These results suggest that the syndrome is manifested during adulthood, which is in accordance to the mean age of the sudden death victims (35 to 40 years). The youngest patient known was 6 months old at the time of sudden death, and the oldest 74.
In the Dutch area of Belgium (6 million inhabitants) already 44 families with the syndrome have been identified. About 15% of members are affected. That gives an incidence of 1:30.000 in that area.
PATHOLOGICAL FINDINGS:
Pathological data are available from many patients with the syndrome. None of the patients have any structural abnormality. Despite other authors suggestions, there is no indication that the disease is arrhythmogenic right ventricular dysplasia. Other non-invasive studies like nuclear magnetic resonance of the heart and echocardiography (which are available in the majority of the patients) are normal.
The genetic defects in Brugada syndrome are not related to the loci of right ventricular dysplasia, and none of the families studied is linked to these loci
CLINICAL MANIFESTATIONS:
The complete syndrome is characterized by episodes of fast polymorphic VT (picture 2)in patients with an electrocardiogram showing a pattern of right bundle branch block and ST segment elevation in leads V1 to V3 (picture3). The manifestations of the syndrome are caused by episodes of polymorphic ventricular tachycardia-ventricular fibrillation. When the episodes terminate spontaneously the patient develops syncopal attacks. When the episodes are sustained, full blown cardiac arrest and eventually sudden death occur. Thus, these manifestations can range widely: at the one end of the spectrum we have asymptomatic individuals and at the other end those who die suddenly. As it is seen in other clinical-electrocardiographic syndromes, there are other different presentations of the disease.
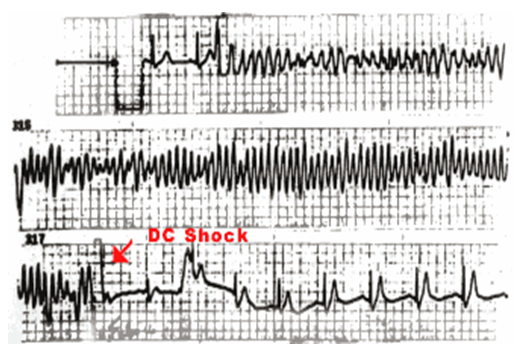
Fig: 1
Spontaneous polymorphic ventricular tachycardia recorded during monitoring in a patient with Brugada syndrome. The arrhythmias are fast and need DC shock to terminate.
There exist asymptomatic individuals in whom the typical electrocardiogram is detected during routine examination. This electrocardiogram cannot be distinguished from that of symptomatic patients. In other patients, the characteristic electrocardiogram is recorded during screening after the sudden death of a family member with the disease. On the other hand, there is the group of symptomatic patients who have been diagnosed as suffering syncopal episodes of unknown cause or vaso-vagal origin, or have a diagnosis of idiopathic ventricular fibrillation. Some of these patients are diagnosed at follow-up, when the electrocardiogram changes spontaneously from normal to the typical pattern of the syndrome (picture 5). This is also the case for those individuals in whom the disease is unmasked by the administration of an antiarrhythmic drug given for other arrhythmias, for instance atrial fibrillation.
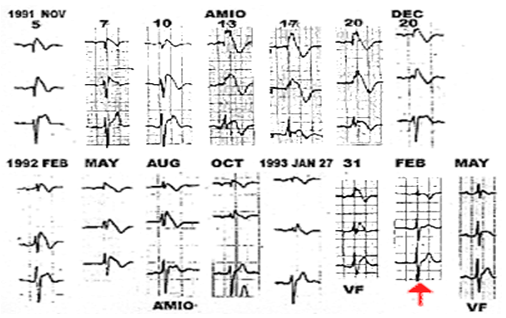
Fig: 2
Spontaneous changes on the electrocardiogram in a patient with Brugada syndrome. Note how the ST elevation changes. On February 1993 the electrocardiogram was completely normal (arrow).
It really does not make a difference, this electrocardiogram, with or without symptoms, appearing spontaneously or after the administration of medications, is a marker of sudden death: Up to 40% of the individuals will develop a new or a first episode of polymorphic ventricular tachycardia or sudden death during the next 2-3 year follow-up. The only excepcion seem to be carriers who are asymptomatic and in whom the electrocardiogram is only found after administration of drugs.
REFERENCES:
1. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992; 20: 1391-1396.
2. Ferracci A, Fromer M, Schlapfer J, Pruvot E, Kappenberger L. Primary ventricular fibrillation and early recurrence: apropos of a case of association of right bundle branch block and persistent ST segment elevation. Arch Mal Coeur Vaiss. 1994; 87: 1359-1362.
3. Proclemer A, Facchin D, Feruglio GA, Nucifora R. Recurrent ventricular fibrillation, right bundle-branch block and persistent ST segment elevation in V1-V3: a new arrhythmia syndrome? A clinical case report. G Ital Cardiol. 1993; 23: 1211-1218.
4. Brugada J, Brugada P. Further characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden cardiac death. J Cardiovasc Electrophysiol. 1997; 8: 325-331.
5. Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: a marker for sudden death in patients without demonstrable structural heart disease. Circulation. 1998; 97: 457-460.
6. Alings M, Wilde A. “Brugada” syndrome: clinical data and suggested pathophysiological mechanism. Circulation. 1999; 99: 666-673.
7. Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation. 2000; 102: 2509-2515.
8. Brugada P, Brugada R, Brugada J. Sudden death in patients and relatives with the syndrome of right bundle branch block, ST segment elevation in the precordial leads V1 to V3 and sudden death. Eur Heart J. 2000; 21: 321-326.
9. Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998; 392: 293-296.
10. Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999; 85: 803-809.
11. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999; 100: 1660-1666.
12. Rook MB, Bezzina Alshinawi C, Groenewegen WA, et al. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999; 44: 507-517.
13. Deschenes I, Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000; 46: 55-65.
14. Wilde AAM, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome. Eur Heart J. 2002; 23: 1648-1654.
15. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005; 111: 659-670.
16. Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. 2006; 29: 1130-1159.
17. Miyasaka Y, Tsuji H, Yamada K, et al. Prevalence and mortality of the Brugada-type electrocardiogram in one city in Japan. J Am Coll Cardiol. 2001; 38: 771-774.
18. Donohue D, Tehrani F, Jamehdor R, Lam C, Movahed MR. The prevalence of Brugada ECG in adult patients in a large university hospital in the western United States. Am Heart Hosp J. 2008; 6: 48-50.
19. Hermida JS, Lemoine JL, Aoun FB, Jarry G, Rey JL, Quiret JC. Prevalence of the Brugada syndrome in an apparently healthy population. Am J Cardiol. 2000; 86: 91-94.
20. Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002; 11: 337-345.
21. Schulze-Bahr E, Eckardt L, Paul M, Wichter T, Breithardt G. Molecular genetics of the Brugada syndrome. In: Antzelevitch C, Brugada P, editors. The Brugada Syndrome: from Bench to Bedside. 1st ed. Blackwell Publishing; 2005. p. 42-51.
22. Vatta M, Dumaine R, Antzelevitch C, et al. Novel mutations in domain I of SCN5A cause Brugada syndrome. Mol Genet Metab. 2002; 75: 317-324.
23. Pfahnl AE, Viswanathan PC, Weiss R, et al. A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm. 2007; 4: 46-53.
24. Casini S, Tan HL, Bhuiyan ZA, et al. Characterization of a novel SCN5A mutation associated with Brugada syndrome reveals involvement of DIIIS4-S5 linker in slow inactivation. Cardiovasc Res. 2007; 76: 418-429.
25. Cordeiro JM, Barajas-Martinez H, Hong K, et al. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation. 2006; 114: 2026-2033.
26. Schulze-Bahr E, Eckardt L, Breithardt G, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat. 2003; 2: 651-652.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








