

 About Authors:
About Authors:
Sreedhar Reddy C
M.Pharm, Department of Pharmaceutics,
Vasavi Insitute of Pharmaceutical Sciences,
Vasavi Nagar, Peddapalli (V), Kadapa (dist),
A.P, India
Abstract:
In this study, transdermal patches containing Itraconazole were prepared using different ratios of polyvinylpyrrolidone (PVP) and Hydroxy propyl methyl cellulose (HPMC) by solvent evaporation technique using 10%w/w of dibutyl phthalate incorporated as plasticizer. The drug matrix film of PVP and HPMC was casted on a polyvinylalcohol backing membrane that was previously dried at 600C for 6 hrs. All the prepared formulations were subjected to physical studies (moisture content, moisture uptake, Tensile strength, flatness and Drug content determination), in vitro release studies and in vitro skin permeation studies. The physiochemical compatibility of the drug and the polymers studied by IR spectroscopy have absence of any incompatibility. In vitro permeation studies were performed across skin using a Franz diffusion cell. Variations in drug release profiles among the formulations studied were observed. Based on a physicochemical and in vitro skin permeation study, formulation F1 (PVP/HPMC, 5:1) and F5 (PVP/HPMC, 1:5) were chosen for further in vivo experiments. The anti inflammatory effect and a sustaining action of Itraconazole from the two transdermal patches selected were studied by inducing paw edema in rats with 1% w/v carrageenan solution. Hence, it can be reasonably concluded that Itraconazole can be formulated into the transdermal matrix type patches to sustain its release characteristics.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1247
INTRODUCTION
Transdermal drug delivery systems (TDDS) are adhesive drug-containing devices of defined surface area that delivers a predetermined amount of drug to the intact skin at a preprogrammed rate1. The transdermal delivery has gained importance in recent years.Transdermal delivery of drugs is a novel drug delivery system and this system breaks many barriers in drug therapy like need of assistance, intermediate dosing and uncomfortable administration. Transdermal delivery has many advantages over conventional modes of drug administration, it avoids hepatic first pass metabolism, potentially decreases side effects and improves patient compliance. FDA approved the first transdermal patch products in 1981.
Fungal infection of skin is now a days one of the common dermatological problems. The physicians have a wide choice for treatment from transdermal and to liquid formulations. Amongst the topical transdermal formulations have been widely accepted in both cosmetics and pharmaceuticals2.
Transdermal therapeutics systems are defined as self contained discrete dosage forms when applied to the intact skin, deliver the drugs through the skin, at controlled rate to the systemic circulation. The advantages of delivering drugs across the skin for systemic therapy are well documented. Some of the main advantages of a transdermal drug delivery system are:
The simplified medication regimen leads to improved patient compliance and reduced inter and intra patient variability.
1. Self administration is possible with these systems.
2. The drug input can be terminated at any point of time by removing transdermal patch.
3. These advantages are however counter-balanced by a umber of limitations.
These include the following Skin irritation or contact dermattis due to the drug excipients and enhancer of the drug used to increase percutaneous absorption is another limitation3
1. The barrier function of the skin changes from one site to another on the same person, from person to erson and with age.
2. Clinical need is another area that has to be examined carefully before a decision is made to develop a transdermal product.
Itraconazole is a newer water soluble triazole antifungal drug used for the treatment of supericial fungal infections4. The mechanism of action of triazoles they inhibit the fungal cytocheome P450 enzyme lanosterol 14-demethylas and thus impair ergosterol synthesis leading to a cascade of membrane abnormalities in the fungus. The lower toxicity of triazoles compared to imidazoles has correlated with their lower affinity for mammalian CYP450 and lesser propensity to inhibit mammalian sterol synthesis. It is available as tablets for oral administration, as a powder for oral suspension and as a sterile solution for intravenous use. It is widely used in vaginal candidias, oropharyngeal and esophageeal candidiasis and cryptococccal meningitis. It is also effective for the tretment of candida urinary tract infections peritonitis and systemic candida infections including candidemia, and pneumonia
MATERIALS AND METHODS
Itraconazole was received as a gift samples from Dr. Reddy’s Laboratory Hyderabad; Polyvinylpyrrolidone (PVP), Hydroxy Propyl Methyl cellulose (HPMC) and Oleic acid were obtained from SD Fine chemical Ltd, Mumbai. Dibutylphthalate was obtained from Sigma chemicals Ltd Ahmedabad, India. Chloroform, methanol, potassium, glycerol, potassium dihydrogen phosphate, etc. were of analytical grade. Double-distilled water was used throughout the study.
INVESTIGATION OF PHYSICOCHEMICAL COMPATIBILITY OF DRUG AND POLYMER
The physicochemical compatibility between Itraconazole and polymers used in the films was studied by using Fourier transform infrared (FTIR - 8300, Shimadzu Co., Japan) spectroscopy5. The infrared (IR) spectra were recorded using an FTIR by the KBr pellet method and spectra were recorded in the wavelength region between 4000 and 400 cm–1. The spectra obtained for ITZ, polymers, and physical mixtures of ITZ with polymers were compared.
[adsense:468x15:2204050025]
PREPARATION OF TRANSDERMAL FILMS
Transdermal patches containing ITZ were prepared by the solvent evaporation technique in cylindrical glass molds with both sides opens. The backing membrane was cast by pouring a 2 % (m/V) polyvinyl alcohol (PVA) solution followed by drying at 60°C for 6 h. The drug reservoir was prepared by dissolving PVP and HPMC in Chloroform. The ratio of polymers were varied for all the formulation keeping the total weight fixed at 150mg. Dibutyl phthalate 15 % (w/w of dry polymer composition) was added as a plasticizer6. The drug Itraconazole was added into the homogeneous dispersion under slow stirring with a magnetic stirrer. The uniform dispersion was cast on a PVA backing membrane and dried at room temperature. (Table 1) The films were kept in desiccator for further study.
Table 1: Composition of Transdermal patches
|
Ingredients |
Formulation code |
||||
|
ITZ 1 |
ITZ 2 |
ITZ 3 |
ITZ 4 |
ITZ5 |
|
|
Itraconazole (mg) |
70 |
70 |
70 |
70 |
70 |
|
PVP (mg) |
130 |
115 |
80 |
65 |
50 |
|
HPMC (mg) |
50 |
65 |
80 |
115 |
130 |
|
Dibutylphthalate % |
15 |
15 |
15 |
15 |
15 |
|
Oleic acid (ml) |
0.25 |
0.25 |
0.25 |
0.25 |
0.25 |
|
Chloroform (ml) |
10 |
10 |
10 |
10 |
10 |
PHYSICOCHEMICAL EVALUATION OF FILMS
Thickness of the patch7 The thickness of patches was measured at three different places using a micrometer (Mitutoyo Co., Japan) and mean values were calculated.
Weight Variation8 The patches were subjected to mass variation by individually weighing randomly selected patches. Such determination was carried out for each formulation.
Moisture content9 The patches (n =3) were weighed individually and kept in a desiccator containing calcium chloride at 37oC for 24 hrs. The final weight was noted when there was no change in the weight of individual patch. The percentage of moisture content was calculated as a difference between initial and final weight with respect to final weight.
Moisture uptake10 A weighed film kept in desiccators at 40oC for 24h was taken out and exposed to two different relative humidity of 75%RH (saturated solution of sodium chloride) and 93%RH (saturated solution of ammonium hydrogen phosphate) in two different desiccators respectively at room temperature then the weights were measured periodically to constant weights.
Flatness Longitudinal strips were cut out from the prepared medicated film the lengths of each strip were measured11. Then variation in the length due to the non-uniformity in flatness was measured. Flatness was calculated by measuring constriction of strips and a zero percent constriction was considered to be equal to a hundred percent flatness.
Constriction (%) = L1-L2 × 100
L2
Where,
L1- initial length of strip
L2 - final length of strip.
Determination of tensile strength
In order to determine the elongation as a tensile strength, the polymeric patch was pulled by means of a pulley system weights were gradually added to the pan to increase the pulling force till the patch was broken12. The elongation i.e. the distance traveled by the pointer before break of the patch was noted with the help of magnifying glass on the graph paper, the tensile strength was calculated as kg cm-2.
Folding Endurance 12 This was determined by repeatedly folding one film at the same place till it broke. The number of times the film could be folded at the same place without breaking gave the value of folding endurance.
Water vapour transmission (WVT) rate WVTR is defined as the quantity of moisture transmitted through unit area of film in unit time. The film was fixed over the brim of a glass vial, containing 3 g of fused calcium chloride as desiccant, with an adhesive tape13. The vial was weighed and kept in desiccators containing saturated solution of potassium chloride to provide relative humidity of 84%. The vial was taken out and weighed at every 24 hrs interval for a period of 72 hrs. The water vapour transmission rate was calculated from the plots of amount of water vapour transmitted versus time.
Drug content determination The patches at 1cm2 were cut and added to a beaker containing 100ml of Phosphate buffered solution of pH 7.4. The medium was stirred with a Teflon coated magnetic bead for 5hrs14. The solution was later filtered and analyzed for drug content with proper dilution at 276 nm spectrophotometrically
In-vitro drug release studies14
The fabricated film was placed on the rat skin and attached to the diffusion cell such that the cell’s drug releasing surface towards the receptor compartment which was filled with phosphate buffer solution of pH 7.4 at 37±10C. The elution medium was stirred magnetically. The aliquots (5ml) were withdrawn at predetermined time intervals and replaced with same volume of phosphate buffer of pH 7.4. The samples were analyzed for drug content using UV spectrophotometer at 276 nm.
Kinetics of drug release15
To examine the drug release kinetics and mechanism, the cumulative release data were fitted to models representing zero order (Q v/s t), first order [Log (Q0-Q) v/s t], Higuchi’s square root of time (Q v/s t1/2) and Korsemeyer Peppas double log plot (log Q v/s log t) respectively, where Q is the cumulative percentage of drug released at time t and (Q0-Q) is thecumulative percentage of drug remaining after time t.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
IN-VIVO STUDIES
a. Anti-inflammatory studies
The animals, wistar rats were divided into two groups16. Acute inflammation was produced by sub planter injection 0.1ml of 1% solution of carrageenan in normal saline, in the right hind paw of the rats; one hour after application of patch .The paw volume was measured by using plethysmometer at regular periods of time interval after the injection of carrageenan. The difference between the two readings was taken as the volume of the edema and the percentage anti inflammatory activity was calculated by using the following formula
RESULTS AND DISCUSSION
Investigation of Physicochemical Compatibility of Drug and Polymer
Drug & excipients interactions play a vital role with respect to release of drug from the formulation amongst others. FTIR techniques have been used here to study the physical and chemical interaction between drug and excipients used. Infrared absorption spectroscopy (IR)
For Itraconazole 2950 C-H cm-1 stretching, 1280 C-F cm-1 stretching, 1460C-H cm-1bending,For HPMC & Itraconazole 2950 C-H cm-1 stretching, 1460 C-H cm-1 bending C-F bond is not found as shown in Fig. 2, For PVP & Itraconazole 2960 C-H Stretching-g Ar, 1460, C-C Multiple bond stretching (Aromatic), which show there were no physical interactions because of some bond formation between drug and polymers. (Figure 1, 2, 3)
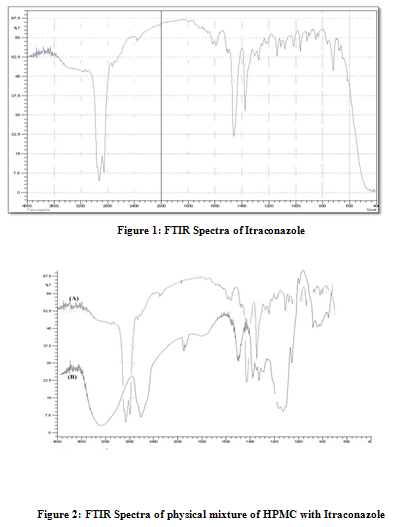
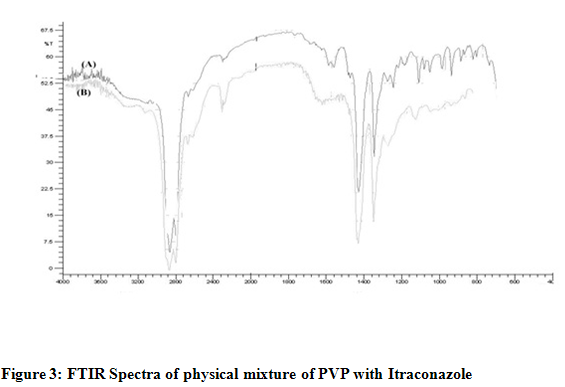
Physicochemical evaluation of films
The results of the physicochemical evaluation of the patches are shown in Table 2. The thickness ranged between 0.16 to 0.21 ± 0.01 mm, which indicates that they are uniform in thickness. The weights ranged between 160 ± 3.1mg to 220 ± 2.5 mg, which indicates that different batches patch weights, were relatively similar. Good uniformity of drug content among the various batches was observed, with all formulations and ranged from 96.9 ± 0.2 % to 98.3 ± 0.2 %. The results indicate that the process employed to prepare patches in this study was capable of producing patches with uniform drug content and minimal patch variability. The flatness study showed that all the formulations had the same strip length before and after their cuts, indicating 99% flatness. Thus, no amount of constriction was observed; all patches had a smooth, flat surface; and that smooth surface could be maintained when the patch was applied to the skin. Folding endurance test results indicated that the patches would not break and would maintain their integrity with general skin folding when applied. Moisture content and moisture uptake studies indicated that the increase in the concentration of hydrophilic polymer was directly proportional to the increase in moisture content and moisture uptake of the patches. The moisture content of the prepared formulations was low, which could help the formulations remain stable and reduce brittleness during long termstorage. The moisture uptake of the formulations was also low, which could protect the formulations from microbial contamination and reduces bulkiness.
Table 2: Evaluation of Transdermal films
|
Parameters |
ITZ 1 |
ITZ 2 |
ITZ 3 |
ITZ 4 |
ITZ 5 |
|
Weight variation |
210±1 |
180±2.10 |
160±3.1 |
220±2.5 |
200±4.0 |
|
Thick ness |
0.19±0.02 |
0.21±0.01 |
0.18±0.01 |
0.20±0.01 |
0.16±0.01 |
|
Drug Content |
98.2±0.1 |
97.1±0.3 |
96.9±0.2 |
97.8±0.3 |
98.3±0.2 |
|
Flatness |
99% |
99% |
99% |
99% |
99% |
|
Tensile strength |
12.13±2.12 |
12.55±1.67 |
12.89±1.89 |
13.23±1.34 |
13.23±1.98 |
|
Folding endurance |
160.2±4.20 |
180±3.45 |
210±3.23 |
234±6.7 |
254.2±5.6 |
|
WVTR |
4.120±0.553 |
3.143±0.436 |
4.111±0.254 |
3.189±0.06 |
2.50±0.45 |
|
Moisture content |
4.115±0.05 |
3.823±0.23 |
3.542±0.09 |
3.234±0.07 |
2.987±0.03 |
Table 3: Kinetic modeling of drug release
|
Model |
Equation |
ITZ F1 |
ITZ F2 |
ITZ F3 |
ITZ F4 |
ITZ F5 |
|
Zero order |
Mo-Mt=kt
|
0.981 |
0.812 |
0.915 |
0.985 |
0.990 |
|
First order |
InM=InMo |
0.985
|
0.830 |
0.916 |
0.799 |
0.867 |
|
Higuchi’s Matrix |
M0−Mt = kt1/2 |
0.850 |
0.835 |
0.930 |
0.988 |
0.900 |
|
Korsmeyer-Peppar |
log (M0-Mt)= log k + n logt |
0.890 |
0.840 |
0.940 |
0.982 |
0.970 |
|
Hixon crowell |
M01/3−Mt1/3 |
0.905 |
0.845 |
0.918 |
0.938 |
0.950 |
In-vitro skin permeation
The in-vitro release profile is an important tool that predicts in advance how a drug will behave in vivo. The results of in-vitro skin permeation studies of ITZ from transdermal patches are shown in Figures 5. In the present study combination of hydrophilic (PVP) and hydrophobic (HPMC) polymers are used to prepared patches. Formulation ITZF1 exhibited greatest 80.12 % of drug release value, while formulation ITZF5 exhibit lowest 50.34% of drug release value.
Anti-inflammatory activity
Anti-inflammatory activity was performed for ITZF1 and compared with ITZF5 formulations. ITZF1 produced 91.04% inhibition of paw edema in rats 10 hrs after carrageenan insult, whereas in the case of formulation ITZF5, the value became 43.34% at 10 hrs after the carrageenan insult. The percentage inhibition of ITZF1 was 2.1 folds greater than F5.
The cumulative amount of drug released from formulations containing more amount of hydrophilic polymer and less Hydrophilic polymer in the same formulation, release drug at faster rate than more amount of hydrophobic polymer and less hydrophilic. The cumulative amount of drug released from formulations ITZF1 and ITZF2 is much higher than formulation ITZF3, ITZ F4 and ITZF5. The drug release from the patch is ordered as ITZF1 > ITZF2 > ITZF3 > ITZF4 > ITZF5. Unlike the formulations ITZF2, ITZF3, ITZF4, and ITZF5 and, the formulations F1 achieved a high cumulative amount of drug permeation at the end of 12 hours. Based on physiochemical and in-vitro release experiments, ITZF1 was chosen for further pharmacodynamic studies.
STABILITY STUDY OF OPTIMIZED FORMULATION
Stability study was carried out for optimized patch (F1) formulation at 40oC temperature in a humidity chamber having 75 % RH for 3 months. After 3 months samples were withdrawn and evaluated for physicochemical properties and in-vitro diffusion study, which shows no change. (Figure 5)
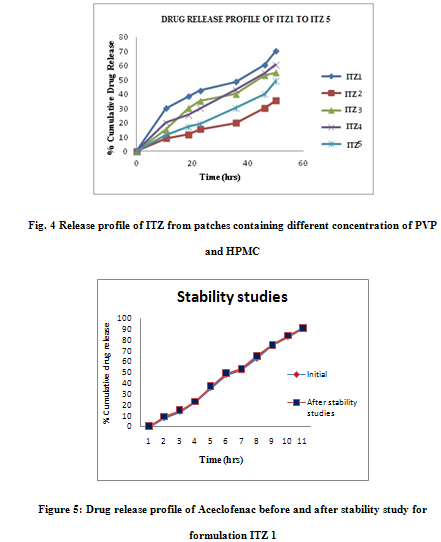
CONCLUSION
The drug release through the transdermal patches of Itraconazole follows First order kinetics with diffusion controlled mechanism. Effect of penetration enhancer like oleic acid has been checked on in-vitro permeation of drug and was found to be effective. The anti inflammatory activity carried out on wistar rats showed that % inhibition of paw edema in rats was good in F1 and skin irritation test performed on albino rabbits showed no signs of redness or erythma. The finding of this result revealed that the problems of Itraconazole on oral administration like dissolution rate limited absorption and gastric side effects can be overcome by applying Itraconazole topically in the form of transdermal patch.
REFERENCES
1. Zhao, K. and Singh, J. Mechanism(s) of in vitro percutaneous absorption enhancers of tamoxifen by enhancers J. Pharm. Sci. 89, 771–780 (2000).
2. Valenta, C., Claders, J., O’Shea, P., and Hardgraft, J. Effect of phloretin on the percutaneous absorption of lignocaine across human skin. J. Pharm. Sci. 90, 485-492 (2001).
3. Morgan, T.M., O’Sullivan, H.J.M., Reed, B.L., and Fainin, B.C. Transdermal delivery of estradiol in postmenopausal women with a novel topical aerosol. J. Pharm. Sci. 87, 1226–1228 (1998).
4. Essential of Medicinal Pharmacology, Fifth Edition 2003, Author. KD.Tripathi, Published by Jaypee brothers, Medical Publishers (P) LTD, New Delhi.
5. Sharna, A., Kara, M., Smith, F.R., Krishnan, T.R. Transdermal drug delivery using electoporation I. Factors inITZencing in vitro delivery of terazosin HCl in hairless rats. J. Pharm. Sci. 89, 528–535 (2000).
6. Tang, H., Blankschtein, D, and Langer, R. Effects of low-frequency ultrasound on the transdermal penetration of mannitol: Comparative studies with in vivo and in vitro skin. J. Pharm. Sci. 91, 1776–1794 (2002).
7. BioElectronics launches ActiPatch Therapy dermal patches. Espicom Business Intelligence une 11, 2004.
8. Banga AK, Bose S, Ghosh TK. Iontophoresis and electroporation: comparisons and contrasts. Int J Pharm 1999; 179:1-19.
9. Guy RH, Kalia YN, Delgado-Charro MB, Merino V, López A, Marro D. Iontophoresis: electrorepulsion and electroosmosis. J Control Rel 2000: 64:129-132.
10. M. Kanebako, T. Inagi, K. Takayama. Transdermal delivery of indomethacin by iontophoresis. Biol. Pharm. Bull., 2002, 25: 779-82.
11. S. Miyazaki, H. Mizuoka, Y. Kohata, et al. External control of drug release and penetration. VI. Enhancing effect of ultrasound on the transdermal absorption of indomethacin from an ointment in rats. Chem. Pharm. Bull., 1992, 40: 2826-2830.
12. J. Asano, F. Suisha, M. Takada, et al. Effect of pulsed output ultrasound on the transdermal absorption of indomethacin from an ointment in rats. Biol. Pharm. Bull., 1997, 20: 288-291.
13. P. Rama Rao, P.V. Diwan. Formulation and evaluation of polymeric films of indomethacin for transdermal administration. Indian J.Pharm. Sci., 1998, 60: 169-171.
14. Y. Chen, P. Wang, X. F. Wang, et al. Study on formulation designing of transdermal delivery system of indomethacin. Chinese J. Hospital Pharmacy, 1999, 8: 451-453.
15. Y. W. Chien, H. Xu, C. C. Chiang, et al. Transdermal Controlled Administration of Indomethacin. I. Enhancement of Skin Permeability. Pharm. Res., 1988, 2: 103-106.
16. N. Kanikkannan, S. B. Jayaswal, J. Singh. Transdermal delivery of indomethacin: II. Effect of penetration enhancers on the in vitro percutaneous absorption from patch formulations. Pharmazie, 1994, 8: 619-620.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






