
ABOUT AUTHORS:
Shikha Attri, Arti Choudhary, Devendra Gupta
M.Pharm in Pharmaceutical Chemistry, Lachoo
Memorial College of Science and Technology,
Jodhpur University, Rajasthan University Of Health Sciences, Jaipur
*shikhu921@gmail.com
ABSTRACT
Thalidomide, removed from clinical use because of severe teratogenicity, is back. In a comeback that has proceeded with remarkable speed, the drug that adversely affected more than 10,000 infants just over four decades ago now seems to be a lifesaver for patients with advanced plasma cell malignancies. Thalidomide and its immunomodulatory (IMiDs) analogs (lenalidomide, CC-4047, ACTIMID) are a novel class of compounds with numerous effects on the body’s immune system, some of which are thought to mediate the anticancer and anti- inflammatory results observed in humans. Thalidomide is currently being used experimentally to treat various cancers and inflammatory diseases. Immunomodulatory activities along with anti-angiogenic, anti-proliferative,and pro-apoptotic properties are thought to mediate the IMiDs’antitumor responses observed in relapsed and refractory multiple myeloma and some solid tumor cancers. This has led to their use in various oncology clinical trials.
A review is presented of the history of thalidomide and its analogues properties with an emphasis on applications in malignant disease.
REFERENCE ID: PHARMATUTOR-ART-1830
1.INTRODUCTION
Thalidomide caused severe malformations in babies born to mothers taking the drug for morning sickness in the late 1950s and early 1960s. It is now known that these teratogenic effects are due to potent anti-angiogenic and immunomodulatory actions. These properties have lead to the testing of thalidomide in a number of infective, inflammatory and malignant conditions. Promising activity has been reported in myeloma, AIDS-related Kaposi's sarcoma, renal cell carcinoma and glioblastoma multiforme.
Thalidomide(Thal) is a derivative of glutamic acid and is pharmacologically classified as an immunomodulatory agent . Structurally, thalidomide contains 2 amide rings and a single chiral center, and its full chemical name is alpha-N{phthalimido}glutarimide [C13H10N2O4] with a gram molecular weight of 258.2. The currently available formulation is a non-polar racemic mixture present as the optically active S and R isomers at physiologic pH, which can effectively cross cell membranes.[1]The S isomer has been linked to thalidomide’s teratogenic effects, whereas the R isomer appears to be primarily responsible for its sedative properties. The isomers rapidly interconvert at physiologic pH in vivo, and thus efforts at formulating only the R isomer have failed to obviate the teratogenic potential of thalidomide.[2]
2. HISTORY
Thalidomide was first synthesized in Germany in 1954 from the glutamic acid derivative α-phthaloylisoglutamine and soon thereafter animal studies showed it to be extremely nontoxic. It was erroneously concluded that the purported structural resemblance to the then widely used barbiturates could indicate its potential as a “safe” sedative. A single questionable study in mice was performed to show its sedative-hypnotic effects. Based on this study, human trials were initiated in Germany under the lax pharmaceutical regulatory environment without comprehensive animal toxicology studies. These studies should have included reproductive toxicology in a non-rodent species such as rabbits or monkeys. Thalidomide was found to be an effective sedative and sleep-inducing agent in humans with less potential for overdose compared with the barbiturates. It was approved in Germany in 1957 and subsequently in other countries including the United Kingdom, Canada, and Australia under brand names such as Contergan, Distaval, Talimol, and Kevadon. Thalidomide was also found to be an effective anti-emetic in pregnancy and its use in this group of patients subsequently increased. The error in this presumption of good efficacy with limited toxicity became apparent when reports of deformed babies started appearing from late 1956. By the time it was withdrawn in 1961, ~5000 to 12000 deformed babies (and an unknown number of aborted fetuses) from 46 countries were already born. Thalidomide was never approved in the United States because of the diligence of the Food and Drug Administration (FDA) reviewer Frances Kelsey who requested more information from the petitioning company on the reported peripheral neuritis. The company was not forthcoming and the application was withdrawn. This would have been the end of the drug were it not for subsequent reports of its effectiveness in treating various inflammatory and dermatological conditions such as ENL. [3]
In August 1998, thalidomide (Figure 1) was approved for sale in the United States for the chronic treatment of erythema nodosum leprosum (ENL), a painful inflammatory dermatologic reaction of lepromatous leprosy. This marked the approval of the world’s most controversial drug after it was withdrawn from Europe more than 40 years ago. [3]
3.CANCER TREATMENT
Thalidomide was first studied as an anticancer agent by astute investigators intrigued by its potent teratogenic potential. In 1962, only 4 months after the initial reports of teratogenicity, Woodyatt6 treated a woman with malignant mixed mesodermal tumor of the uterus. The interest in studying thalidomide as an anticancer agent led to at least three clinical trials in the early 1960s, including a study by the Eastern Cooperative Oncology Group. These trials did not show any significant activity, and interest in thalidomide as an anticancer agent diminished greatly.[4]
In 1994 Harvard Professor Robert D'Amato at Boston Children's Hospital discovered that thalidomide was a potent inhibitor of new blood vessel growth (angiogenesis), which is required for tumor growth.[5] He then showed in a rabbit cancer model that thalidomide suppressed tumor growth in animals.[6]He also found that a subset of anti-inflammatory drugs, such as sulindac and dexamethasone, had moderate anti-angiogenic activity. When these anti-inflammatory anti-angiogenic drugs were combined with thalidomide they increased both thalidomide's anti-angiogenic and anti-tumor activity. Based on these discoveries, numerous cancer clinical trials for thalidomide were initiated with and without dexamethasone.[7]
Thalidomide was initially tested in humans as a single agent for the treatment of multiple myeloma due to its anti-angiogenic activity.[8] The early foundation for this work was laid out in a 1993 keynote lecture at the American Society of Hematology by Dr. Folkman when he hypothesized that all blood borne malignancies are angiogenesis dependent based upon his discovery that the levels of the angiogenic growth factor FGF were elevated in the urine of patients with leukemia.[9] Further studies in his lab showed efficacy with the angiogenesis inhibitor TNP-470 in mouse models of leukemia. Additionally, in 1994 Vacca had shown a five fold increase in angiogenesis in the bone marrow of multiple myeloma patients.[10] When the family of a patient with late stage multiple myeloma requested any possible help from Dr. Folkman in 1997, he attempted to obtain TNP-470 as a therapy. However TNP-470 could not be obtained outside of the ongoing clinical trial and thus Dr. D'Amato suggested that thalidomide be used instead for this patient. A small study was then initiated with thalidomide for this patient and several others by Dr. Bart Barlogie with dramatic effects.[11]
Since then, many studies have shown that thalidomide, in combination with dexamethasone, has increased the survival of multiple myeloma patients.
In 2006 the U.S. Food and Drug Administration granted accelerated approval for thalidomide in combination with dexamethasone for the treatment of newly diagnosed multiple myeloma patients.[12]
Thalidomide appears to inhibit disease progression in multiple myeloma by several mechanisms:
- Inhibition of angiogenesis in the bone marrow, which is needed for myeloma cell proliferation.
- Inhibition of the production of interleukin-6 (IL?6), which is a growth factor for the proliferation of myeloma cells.[13]
- Activation of apoptotic pathways through caspase 8-mediated cell death[13]
- At the mitochondrial level, thalidomide results in induction of c-jun terminal kinase (JNK).[13]
- Activation of T cells to produce IL?2, thereby altering the amount and function of natural killer cells (NK cells), thus augmenting the activity of NK?dependent cytotoxicity.[13]
All physicians prescribing and patients receiving thalidomide must go through the STEPS process to ensure that no more children are born with birth defects traceable to the medication. Celgene has sponsored numerous clinical trials with analogues to thalidomide, such as lenalidomide, that are substantially more powerful and have fewer side effects — except for greater myelosuppression.[14]
The activity of thalidomide in solid tumors is less prominent. Studies in Kaposi’s sarcoma, malignant melanoma, renal cell carcinoma and prostate cancer appear more promising especially when thalidomide is combined with biological agents or with chemotherapy. Limited activity was demonstrated in patients with glioma, while thalidomide appears to be inactive in patients with head and neck cancer, breast or ovarian cancer. In a small trial, Australian researchers found thalidomide caused a doubling of the number of T cells in patients, allowing the patients' own immune system to attack cancer cells.[15]
4. THALIDOMIDE ANALOGUES
The use of angiogenesis inhibitors for the treatment of cancer was first conceptualized over 30 years ago, when Dr. Folkman introduced the idea that angiogenesis is required for continued solid tumor growth [16]. Since then, a number of antiangiogenic agents have emerged for use in cancer therapy. Thalidomide (α -N-phthalimido-glutarimide) has emerged as a potent treatment for several disease entities. The exploration of the antiangiogenic and immunomodulatory activities of thalidomide has led to the study and creation of thalidomide analogues. In 2005 Celgene received FDA approval for lenalidomide (Revlimid) as the first commercially useful derivative. Revlimid is available only in a restricted distribution setting to avoid its use during pregnancy. Further studies are being conducted to find safer compounds with useful qualities. Another more potent analog, pomalidomide, is now FDA approved.[17] These thalidomide analogs can be used to treat different diseases, or used in a regimen to fight two conditions.[18]
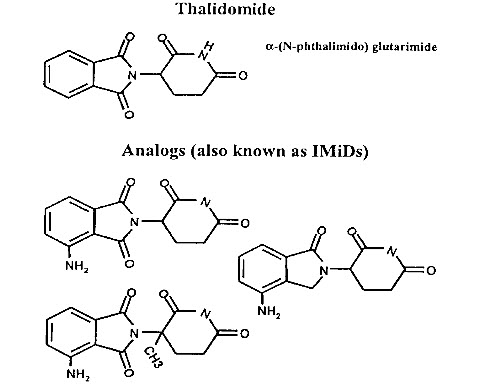
Figure 1. Structures of thalidomide and its potent analogues (immunomodulatory drugs, ImiDs).
4.1 IMiDs – NOVEL THALIDOMIDE ANALOGUES AS ANTICANCER AGENTS
The search for thalidomide analogues with increased immunomodulatory activity and an improved safety profile led to the testing of amino-phthaloyl-substituted thalidomide analogues. These 4-amino analogues, in which an amino group is added to the fourth carbon of the phthaloyl ring of thalidomide Fig.(2), brought about the class termed “IMiDs”. The bioactivities of the IMiDs follow the parent drug thalidomide closely, but with some increase in potency. Fig.(3) summarizes the multifaceted effects of thalidomide and its analogues.
4.1.1. LENALIDOMIDE (CC-5013)
CC-5013, lenalidomide (α -(3-aminophthalimido) glutarimide; Revlimid®)) is an immunomodulatory analogue that has demonstrated higher potency than thalidomide in the HUVEC (human umbilical vein endothelial cells) proliferation and tube formation assays [19]. The proliferation inhibition responded in a dose-dependent manner with increasing concentrations of the drug. Anti-migratory effects as well as tumor growth inhibition in vivo have also been demonstrated [20].
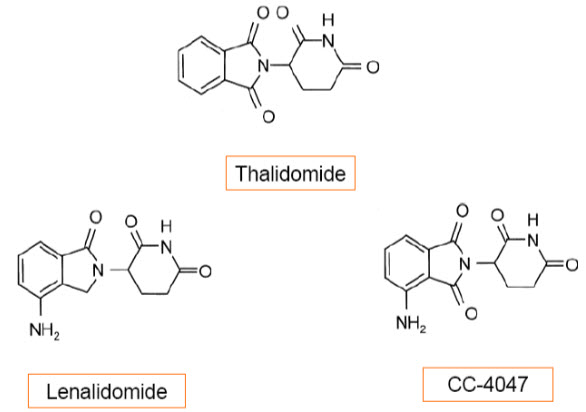
Fig. (2) . Chemical structure of thalidomide, lenalidomide, and CC-4047
4.1.2. ACTIMID (CC-4047)
CC-4047 is a costimulatory thalidomide analogue that can prime protective, long-lasting, tumor-specific, Th1-type responses in vivo [21].
4.1.3. ENMD-0995 (S-3-Amino-phthalimido-glutarimide, S-3APG)
ENMD-0995 is a small molecule analogue of thalidomide that is the S(-) enantiomer 3-amino thalidomide. Thalidomide is a racemic glutamic acid analogue, consisting of S(-) and R(+) enantiomers that interconvert under physiological conditions [22]. The S(-) form potently inhibits release of TNF-alpha from peripheral blood mononuclear blood cells[23], whereas the R(+) form seems to act as a sedative[24]. This 3-amino derivative of thalidomide was demonstrated to have improved angiogenesis inhibitor activity that in animal models has shown no evidence of the toxic side effects as reported for the thalidomide molecule.
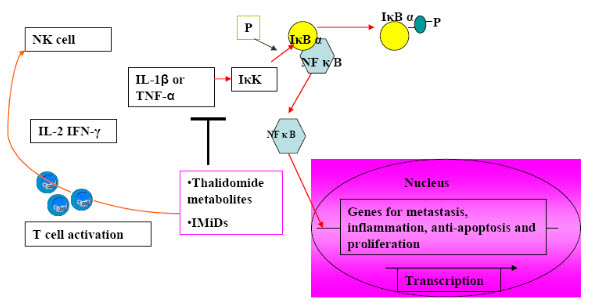
Fig (3). Mechanism of anti-angiogenic and immunomodulatory actions of IMiDsMechanisms of anti-angiogenic and immunomodulatory functions of IMiDs.Immunomodulatory drugs (IMiDs), like thalidomide metabolites upon oxidation, inhibits interleukin 1 β or TNF-alpha – induced activation of IκK, which prevents dissociation of IκBα from NFκB , precluding its nuclear translocation and induction of genes that function in metastasis, angiogenesis, cellular proliferation, inflammation, and protection from apoptosis.IMiDs and thalidomide metabolites also function in T cell activation as its T cell costimulatory function, enhancing T cell proliferation. The activated T cells release interleukin-2 (IL-2) and interferon-gamma (IFN-γ), which activate the Natural Killer cells (NK cells).
5. IMMUNOMODULATORY AND NON-IMMUNOMODULATORY PROPERTIES
The precise mechanism(s) of thalidomide and the IMID®s efficacious activities are unknown. This class of compounds, however, has numerous immunomodulatory and non-immunomodulatory properties, which are probably working in concert to produce the observed efficacy.
Thalidomide and the IMiDs Have Potent Immunodulatory Properties Inhibition of TNF-α, IL-1β, IL-6, IL-12, GM-CSF, and Stimulation of IL-10 in Peripheral Blood Mononuclear Cells Cytokines are soluble glycoproteins released by cells of the immune system that act non-enzymatically through specific receptors to regulate immune responses. TNF-α is a proinflammatory cytokine produced by monocytes, macrophages, lymphocytes, and NK cells. It plays an important role in host immune and inflammatory response to viral, parasitic, fungal, and bacterial infections. TNF-α has been implicated in the pathogenesis of infections and autoimmune diseases. Elevated levels of TNF-α have been associated with various inflammatory and immune disorders such as rheumatoid arthritis, Crohn’s disease, tuberculosis, cancer cachexia, and ENL. Thalidomide’s ameliorative effects on ENL have been particularly striking. Thalidomide and its analogs are potent inhibitors of TNF-α production by lipopolysaccharide-stimulated human monocytes. This inhibition is due to the increased degradation of TNF-α mRNA by thalidomide. Levels of other cytokines, IL-1β, IL-6, and granulocyte macrophage-colony stimulating factor (GMCSF), are also inhibited by thalidomide, whereas IL-10 is stimulated. Lenalidomide and CC-4047 also had similar effects on these cytokines, although with varying degrees of potency compared with thalidomide. The effects of these findings on various diseases are still being investigated.[3]
T Cell Costimulators
Thalidomide and the immunomodulatory drugs induced proliferation of MM patients’ T cells and interleukin-2 and interferon-gamma production in vitro, but were not cytotoxic to MM cells[25]. These drugs enhanced natural killer cell-mediated lysis of autologous tumor cells [26]. The increase in IL2 and IFN-gamma also upregulates the CD40L expression on T cells when IMiDs were added to anti-CD3 stimulated T cells[25]. Although a greater costimulatory effect on the CD8+ T cell subset compared to the CD4+ T cell subset has been observed in one study [27], another showed similar co-stimulation for CD4+ and CD8+ T lymphocytes which correlated with TNFR2 inhibition[28]. This T cell costimulatory effect of the IMiDs may be paradoxical to the suppression of ligand-stimulated release of apoptotic and inflammatory cytokines (i.e., TNF-alpha), and the effects of the IMiDs therefore, similar to thalidomide, results in the clinically divergent effects with regard tospecific pathways involved during an inflammatory response or clinical condition [29].
Pro-Apoptotic Agents
Hideshima et. al. first demonstrated that thalidomide and its analogues induce tumor cell apoptosis, evidenced by increased sub-G1cells or induction of p21 and related G1growth arrest [30]. The IMiDs inhibited the proliferation of chemoresistant MM cells by 20% to 35%, and of Dexamethasone-resistant MM cells by 50%[29]. Enhanced caspase-8 activation, increased sensitivity to Fas induction, reduced expression of cellular inhibitor of apoptosis protein-2, and potentiation of the activities of other apoptosis inducers such as TNF-related apoptosis inducing ligand (TRAIL), has been demonstrated with the IMiDs[31]. This antiproliferative effect was demonstrated in a chromosome 5 deleted (5q-) deletion cell line of Myelodysplasia (MDS) and other hematologic malignancies where the inhibitory concentration of 50% (IC50) was higher for the IMiDs as compared to thalidomide, with induced G0/G1 cell cycle arrest, inhibited Akt and Gab1 phosphorylation, and inhibited the ability of Gab1 to associate with a receptor tyrosine kinase [32].
Anti-Angiogenic Activity
Angiogenesis is the development of new blood vessels. In cancer this can nurture the growth and metastasis of tumors and tumor cells respectively. Thalidomide and IMiDs has been shown to have antiangiogenic properties that are independent of their immunomodulatory effects.This activity is thought to play a role in the apparent efficacy seen with various cancers. In the rat aorta assay, the IMiDs were found to be 2 to 3 times more potent in their antiangiogenic activity compared with thalidomide. Lenalidomide but not thalidomide and CC-4047 significantly inhibited the migration of endothelial cells. CC-4047 on the other hand inhibited VEGF but not basic fibroblast growth factor (bFGF). The IMiDs’ anti-TNF-α activity had no effect on antiangiogenic activity. In multiple myeloma, the close proximity interaction between the indigenous bone marrow stromal cells and patient multiple myeloma cells significantly increased levels of the pro-angiogenic factors VEGF and IL-6 (a multiple myeloma growth and survival factor). Thalidomide and CC-4047 significantly decreased expression of these factors thereby reducing the production and growth of new blood vessels feeding the multiple myeloma cells. These findings underscore the importance of stromal and multiple myeloma cell interaction in the bone marrow microenvironment for the maintenance and progression of the disease and provide another target for thalidomide and its analogs.[3]
ACTIVITY OF THALIDOMIDE AND IMIDS IN MULTIPLE MYELOMA
Thalidomide was serendipitously found to have anti-myeloma activity when it was thought its anti-angiogenic activity could slow the disease by inhibiting the formation of new blood vessels in this highly vascularized cancer. There is now ample evidence to show that the anticancer activity of thalidomide and its analogs in multiple myeloma is through different mechanisms and sites in the bone marrow.[33,34] Fig(4) shows the bone marrow microenvironment in multiple myeloma, which contains aberrations in various cellular processes, immunology, and cell interactions. Thalidomide and the IMiDs’ immunomodulatory activities consist of inhibiting the expression of IL-6 and TNF-α by bone marrow stromal cells that in turn inhibit the growth of multiple myeloma cells. The compounds also enhance T cell stimulation and proliferation with the activated cells, then releasing IL-2 and IFN-γ. These cytokines activate NK cells causing lysis of the multiple myeloma cells.[34] The combination of immunomodulatory and non-immunomodulatory anticancer activities in the bone marrow is thought to produce the significant anti-tumor responses observed in some multiple myeloma patients. This combination activity has significant implications for other blood and solid tumor cancers and is currently being investigated in numerous clinical trials.
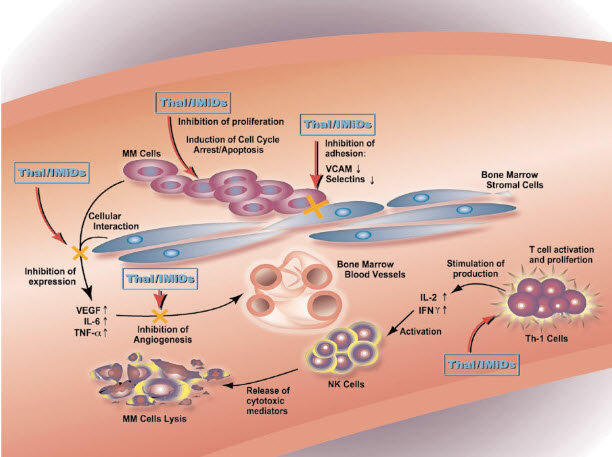
Fig (4).
Sites of activity of thalidomide and IMiDÒs in the bone marrow of multiple myeloma patients.
ACTIVITY OF THALIDOMIDE AND IMIDS IN SOLID TUMORS
Phase II trials of thalidomide have shown potential activity against some solid tumors.[35,36,37] While the mechanism is unclear, it is thought to involve both immunomodulatory and non-immunomodulatory activities. These tumors produce immunologic suppressive factors that prevent priming and activation of CD4+ and CD8+ T cells of the lymph nodes.[3]Other immune cells such as NK and macrophages are also inhibited and are therefore unable to respond to and destroy tumor cells. Thalidomide and the IMiDs’ costimulatory action on primary human T cells enhance antitumor activity mediated by the Th-1 cytokines IL-2 and IFN-γ. The costimulation is thought to overcome the T cell unresponsiveness and prevent the release of suppressive factors, thereby enabling tumor-specific cells to kill tumor cells.[38] Thalidomide and the IMiDs are also thought to costimulate macrophages and NK cells leading to antitumor activity as discussed earlier. Anti-angiogenic and pro-apoptotic activities of thalidomide and the IMiDs are also thought to play a role in the apparent efficacy seen in various highly vascularized solid tumors through inhibition of VEGF and induction of growth arrest respectively.[34]
FUTURE OF THALIDOMIDE AND ITS ANALOGUES
Thalidomide has re-surfaced in the field of oncology, despite its troubled history as a teratogen. The increasing realization of its potential clinical utility in different neoplasms make the study of thalidomide and its analogues very promising.It has yet to be approved for multiple myeloma. Recent reports of potential efficacy in other solid tumors have increased its experimental use and initiation of further clinical trials. The development of the IMiDs is an example of how active research efforts contribute to the synthesis of new thalidomide analogues that provide improved efficacy and/or reducing toxicity. Particulary lenalidomide has significantly higher immunomodulatory and anti-angiogenic potency then thalidomide and also has less side effects then thalidomide. These properties of IMiDsmade them potentially important therapeutics in cancer.While the IMiDs have shown encouraging results in both animal models and have successfully entered clinical trials, efforts are still underway to improve the toxicity profile as well as understanding further and identifying specific molecular targets that would also help delineate the neoplasms for which it may exhibit clinical potency.
CONCLUSION
The rehabilitation of thalidomide has increased its experimental use in numerous oncologic and inflammatory conditions and its analogues also shows a promising result in different malignancies but the exact mechanism of anti tumour activity of thalidomide is unknown. Thalidomide possesses many properties that can help us to explain its anti tumour activity but these are not so specific in explaining about targets. Therefore, despite the success of these agents in certain types of neoplasms, the specific molecular mechanisms and targets are still incompletely understood. Understanding of the precise mechanisms of action will help in the rational design of better thalidomide analogues, optimizing clinical applications, and ultimately translating into beneficial activity in specific neoplasms.
REFERENCES
1.Tseng S, et al.(1996) Rediscovering thalidomide: a review of its mechanism of action, side effects, and potential uses. J Am Acad Dermatol ; 35 : 969-79.
2.Muller GW. Thalidomide: From tragedy to new drug discovery. Chemtech 1997 : 21-215.
3.Steven K. Teo(2005) , “Properties of Thalidomide and its Analogues: Implications for Anticancer Therapy” The AAPS Journal; 7 (1) Article 3 (http://www.aapsj.org).
4.“Thalidomide in the Treatment of Plasma Cell Malignancies”, Journal of Clinical Oncology, Vol 19, No 16 (August 15), 2001: pp 3593-3595
5.D'Amato, R. J.; Loughnan, M. S.; Flynn, E.; Folkman, J. (1994). "Thalidomide is an inhibitor of angiogenesis". Proceedings of the National Academy of Sciences of the United States of America 91 (9): 4082–4085.
6.Verheul, H. M. W.; Panigrahy, D.; Yuan, J.; d’Amato, R. J. (1999). "Combination oral antiangiogenic therapy with thalidomide and sulindac inhibits tumour growth in rabbits". British Journal of Cancer 79 (1): 114–118.
7.vectorblog.org/2013/04/from-thalidomide-to-pomalyst-better-living-through-chemistry/
8.Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E. et al. (1999). "Antitumor Activity of Thalidomide in Refractory Multiple Myeloma". New England Journal of Medicine 341 (21): 1565–1571.
9. ncbi.nlm.nih.gov/pubmed/7508518
10. ncbi.nlm.nih.gov/pubmed/7527645
11. ncbi.nlm.nih.gov/pubmed/10564685
12."FDA Approval for Thalidomide". National Cancer Institute. Retrieved 8 January 2012.
13.Anderson, K. C. (2005). "Lenalidomide and Thalidomide: Mechanisms of Action—Similarities and Differences". Seminars in Hematology 42 (4 Suppl 4): S3–S8.
14.Rao KV (September 2007). "Lenalidomide in the treatment of multiple myeloma". American Journal of Health-system Pharmacy : AJHP : Official Journal of the American Society of Health-System Pharmacists 64 (17): 1799–807.
15.Brown, RD; Spencer A, Ho PJ, Kennedy N, Kabani K, Yang S, Sze DM, Aklilu E, Gibson J, Joshua DE (2009 Nov). "Prognostically significant cytotoxic T cell clones are stimulated after thalidomide therapy in patients with multiple myeloma". Leukemia & Lymphoma 50 (11): 1860–4.
16. Folkman J (1975), “ Tumor angiogenesis: a possible control point in tumor growth”, Ann Intern Med;82:96–100.
17. Search of: pomalidomide". Clinicaltrials.gov. Retrieved 2012-09-01.
18. Raghupathy R, Billett HH (March 2009). "Promising therapies in sickle cell disease". Cardiovasc Hematol Disord Drug Targets 9 (1): 1–8.
19.Tohnya TM, Hwang K, Lepper ER, et al.( 2004), “ Determination of CC-5013, an analogue of thalidomide, in human plasma by liquid chromatography-mass spectrometry”. J Chromatogr B Analyt Technol Biomed Life Sci; 811:135–41.
20.Raje N, Anderson KC (2002), “ Thalidomide and immunomodulatory drugs as cancer therapy”, Curr Opin Oncol; 14:635–40.
21.Dredge K, Marriott JB, Todryk SM, et al.(2002), “ Protective antitumor immunity induced by a costimulatory thalidomide analog in conjunction with whole tumor cell vaccination is mediated by increased Th1- type immunity” . J Immunol; 168:4914–9.
22.Kenyon BM, Browne F, D’Amato RJ (1997 ), “ Effects of thalidomide and related metabolites in a mouse corneal model of neovascularization”. Exp Eye Res ; 64:971–8.
23.Wnendt S, Finkam M, Winter W, Ossig J, Raabe G, Zwingenberger K ( 1996) “Enantioselective inhibition of TNF-alpha release by thalidomide and thalidomide-analogues”. Chirality ; 8:390-6.
24. Frederickson RC, Slater IH, Dusenberry WE, Hewes CR, Jones GT, Moore RA (1977), “A comparison of thalidomide and pentobarbital - new methods for identifying novel hypnotic drugs”. J Pharmacol Exp Ther ; 203:240–51.
25.Corral LG, Haslett PA, Muller GW, et al.( 1999), “ Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha”. J Immunol; 163:380–6.
26.Davies FE, Raje N, Hideshima T, et al.(2001) , “Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma”. Blood ; 98:210–6.
27. Corral LG, Kaplan G ( 1999), “ Immunomodulation by thalidomide and thalidomide analogues”. Ann Rheum Dis ;58(Suppl 1):I107–13.
28.Marriott JB, Clarke IA, Dredge K, Muller G, Stirling D, Dalgleish AG(2002),“Thalidomide and its analogues have distinct and opposing effects on TNF-alpha and TNFR2 during co-stimulation of both CD4(+) and CD8(+) T cells”. Clin Exp Immunol ; 130:75–84.
29.Jeanny B. Aragon-Ching, Haiqing Li, Erin R. Gardner, and William D. Figg(June 2007), “Thalidomide Analogues as Anticancer Drugs” NIH Public Access Author Manuscript ;Recent Pat Anticancer Drug Discov.; 2(2): 167–174.
30.Hideshima T, Chauhan D, Shima Y, et al.( 2000), “ Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy”. Blood;96:2943–50.
31.Mitsiades N, Mitsiades CS, Poulaki V, et al.( 2002), “Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications”. Blood; 99:4525–30.
32. Gandhi AK, Kang J, Naziruddin S, Parton A, Schafer PH, Stirling DI (2006), “Lenalidomide inhibits proliferation of Namalwa CSN.70 cells and interferes with Gab1 phosphorylation and adaptor protein complex assembly”. Leuk Res;30:849–58.
33.Richardson P, Schlossman R, Anderson K (2002), “ Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma”. Blood.;100:3063-3067.
34.Hideshima T, Chauhan D, Shima Y, et al.(2000), “Thalidomide and its analog overcome drug resistance of human multiple myeloma cells to conventional therapy”. Blood.;96:2943-2950.
35.Singhal S, Mehta J, Desikan R, et al.( 1999), “Anti-tumor activity of thalidomide in refractory multiple myeloma”. N Engl J Med ;341:1565-1571.
36.Hwu W, Krown S, Menell J, et al.(2003), “ Phase II study of temozolomide plus thalidomide for the treatment of metastatic melanoma”. J Clin Oncol ;21:3351-3356.
37. Maples W, Stevenson J, Sumrall S, Naughton M, Schwartz J(2004), “Advanced pancreatic cancer: a multi-institutional trial with gemcitabine and thalidomide”. J Clin Oncol ;22:4082.
38.Haslett P, Corral L, Albert A, Kaplan G (1998), “Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation,cytokine production, and cytotoxic responses in the CD8+ subset”. J ExpMed ;187:1885-1982.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








