

ABOUT AUTHORS:
Jain Deepika*, Rathore Kamal Singh
BN Institute of Pharmaceutical Sciences,
Udaipur-Raj.313002 INDIA
*dpka88jain@gmail.com
ABSTRACT
Pharmacovigilance is particularly concerned with adverse drug reactions, or ADRs, which are officially described as: “A response to a drug which is noxious and unintended, and which occurs at doses normally used for the prophylaxis, diagnosis or therapy of disease, or for the modification of physiological function”. The mission of Pharmacovigilance is to contribute to the protection of public health in the regulation of the safety; quality and efficacy of medicines for human use and to ensure the healthcare professionals and patients have access to information about the safe and effective use of medicine. The World Health Organization (WHO) defines an adverse drug reaction (ADR) as “Any response to a drug which is noxious and unintended, and which occurs at doses normally used in man for prophylaxis, diagnosis or therapy of disease or for modification of the physiological function”.
The discipline of pharmacovigilance has developed considerably since the 1972 WHO technical report, and it remains a dynamic clinical and scientific discipline. It has been essential to meet the challenges of the increasing range and potency of medicines (including vaccines), which carry with them an inevitable and sometimes unpredictable potential for harm. The following is a summary of some of the serious challenges facing pharmacovigilance programmes in the next ten years & the major challenges are:Globalization, Web-based sales and information, Broader safety concerns, Public health versus pharmaceutical industry economic growth , Monitoring of established products, Developing and emerging countries, Attitudes and perceptions to benefit and harm, Outcomes and Impact.
REFERENCE ID: PHARMATUTOR-ART-1913
1. Introduction
‘Not all hazards can be known before a drug is marketed’.
- Committee on Safety of Drugs, Annual Report 1969, 1970.
Drug
A pharmaceutical product, used in or on human body for the prevention (prophylaxis), mitigation, diagnosis and/or treatment of disease, or for modification of physiological function.
Medicines have led otiovement in the treatment and control of diseases; they also produce adverse effects on the human body from time to time. Many drugs are precisely targeted to the cause and mechanism of a disease, they may also have minor or distressing effects on other parts of the body, or interact negatively with the various systems of a particular individual or with other drugs or substances that they are taking, or, not work well or at all for some, many or all of those who take them. There is no such thing as a safe drug. There are risks in any intrusion into the human body, whether chemical or surgical. Nothing in this field is entirely predictable - except that the interaction between science and the human body may produce surprises1-3.
Benefits: Benefits are commonly expressed as the proven therapeutic good of a product, but should also include the patient’s subjective assessment of its effects
Risk: Risk is the probability of harm being caused, usually expressed as a percent or ratio of the treated population.
Harm: Harm is the nature and extent of the actual damage that could be caused. It should not be confused with risk.
Effectiveness : Effectiveness is used to express the extent to which a drug works under real world circumstances, i.e., clinical practice (not clinical trials).
Efficacy: Efficacy is used to express the extent to which a drug works under ideal circumstances (i.e. in clinical trials)
Finding the risk of drugs
Pharmaceutical companies are required by law in all countries to perform clinical trials, testing new drugs on people before they are made generally available. The manufacturers or their agents usually select a representative sample of patients for whom the drug is designed, at most a few thousand along with a comparable control group. The control group may receive a placebo and/or another drug that is already marketed for the disease. The purpose of clinical trials is to discover:
- If a drug works and how well
- If it has any harmful effects, and
- Its benefit-harm-risk profile - does it do more good than harm, and how much more? If it has a potential for harm, how probable and how serious is the harm?
Clinical trials in general, tell us a good deal about how well a drug works and what potential harm of the drug, it may cause. They provide information which should be reliable for larger populations with the same characteristics as the trial group - age, gender, state of health, ethnic origin, and so on.
The variables in a clinical trial are specified and controlled and the results relate only to the population of which the trial group is a representative sample. A clinical trial can never tell you the whole story of the effects of a drug in all situations. In fact, there is nothing that could tell you the whole story, but a clinical trial must tell you enough; "enough" being determined by legislation and by contemporary judgments about the acceptable balance of benefit and harm4-9.
Pharmacovigilance
Parmakon(Greek), “drug” and vigilare (Latin), “to keep awake or alert, to keep watch.”
Pharmacovigilance is the pharmacological science relating to the detection, assessment, understanding and prevention of adverse effects, particularly long term and short term side effects of medicines. Generally speaking, pharmacovigilance is the science of collecting, monitoring, researching, assessing and evaluating information from healthcare providers and patients on the adverse effects of medications, biological products, herbalism and traditional medicines with a view to1,10-14:
- Identifying new information about hazards associated with medicines
- Preventing harm to patients
Pharmacovigilance is particularly concerned with adverse drug reactions, or ADRs, which are officially described as: “A response to a drug which is noxious and unintended, and which occurs at doses normally used for the prophylaxis, diagnosis or therapy of disease, or for the modification of physiological function”. Pharmacovigilance is gaining importance for doctors and scientists as the number of stories in the mass media of drug recalls increases15-17.
Because clinical trials involve several thousand patients at most; less common side effects and ADRs are often unknown at the time a drug enters the market. Even very severe ADRs, such as liver damage, are often undetected because study populations are small. Post marketing pharmacovigilance uses tools such as data mining and investigation of case reports to identify the relationships between drugs and ADRs18.
Drug surveillance-WHO
The world health organization (WHO) collects data from 72 countries around the world in an attempt to detect adverse drug reactions (ADRs) signals that are too weak for any individual country to detect. Some of the services provide to the national centers includes-identifying new signals from the information submitted by the national centers, provision of the WHO database, information exchange between national centers and WHO, publications of the newsletters and reports, provision of training and support to the national centers, and annual meeting of the representatives of national centers19.
2. Brief history of Pharmacovigilance in India
Even though pharmacovigilance is still in its infancy, it is not new to India. It was not until 1986 that a formal adverse drug reaction (ADR) monitoring system consisting of 12 regional centers, each covering a population of 50 million, was proposed for India. However, nothing much happened until a decade later when in 1997, India joined the World Health Organization (WHO) Adverse Drug Reaction Monitoring Programmed based in Uppsala, Sweden. Three centers for ADR monitoring were identified, mainly based in teaching hospitals: a National Pharmacovigilance Centre located in the Department of Pharmacology, All India Institute of Medical Sciences (AIIMS), New Delhi and two WHO special centers in Mumbai (KEM Hospital) and Aligarh (JLN Hospital, Aligarh Muslim University). These centers were to report ADRs to the drug regulatory authority of India. The major role of these centers was to monitor ADRs to medicines marketed in India. However, they hardly functioned as information about the need to report ADRs and about the functions of these monitoring centers were yet to reach the prescribers and there was lack of funding from the government. This attempt was unsuccessful and hence, again from the 1st of January 2005, the WHO sponsored and World Bank-funded National Pharmacovigilance Program for India was made operational5, 20-23.
The National Pharmacovigilance Program established in January 2005, was to be overseen by the National Pharmacovigilance Advisory Committee based in the Central Drugs Standard Control Organization (CDSCO), New Delhi. Two zonal centers-the South-West zonal centre (located in the Department of Clinical Pharmacology, Seth GS Medical College and KEM Hospital, Mumbai) and the North-East zonal centre (located in the Department of Pharmacology, AIIMS, New Delhi), were to collate information from all over the country and send it to the Committee as well as to the Uppsala Monitoring centre in Sweden. Three regional centers would report to the Mumbai center and two to the New Delhi one. Each regional center in turn would have several peripheral centers reporting to it. Presently there are 24 peripheral centers24.
Objectives of National Pharmacovigilance Programme
The main objectives of National Pharmacovigilance Programme are as following5
- To foster the culture of Adverse Event (AE) notification and reporting
- To establish a viable and broad based ADR monitoring programs in India
- To create an ADR database for the Indian population
- To create awareness of ADR monitoring among people
- To ensure optimum safety of drug products in Indian market.
- To create infrastructure for ongoing regulatory review of Periodic Safety Updates Regulators (PSURs).
3. Objectives
The mission of Pharmacovigilance is to contribute to the protection of public health in the regulation of the safety, quality and efficacy of medicines for human use and to ensure the healthcare professionals and patients have assess to information about the safe and effective use of medicine25.
Our current ADR reporting system involves a passive approach in which providers are encouraged to report adverse events via e-mail, telephone, or paper. In addition, practitioners may report ADRs directly to a hospital pharmacist who is present in the hospital.
Primary objectives
- Raise awareness on the magnitude of drug safety problems and increase the rational and safe use of drugs.
- Identification of potential safety hazards, evaluation, monitoring and where appropriate, implementation of regulatory action to maximize benefit and minimize risks associated with medicinal products.
- Early detection of unknown adverse drug reaction and increase in frequency of known adverse drug reactions
- To compare the number of ADRs identified by this traditional passive method
- To determine the significance of ADRs those were reported by patients but unknown to their provider.
- Convince healthcare professionals that reporting to ADR is their professional and moral obligation.
- Identification of risk factors and possible mechanism underline adverse reactions
Secondary objectives
- Aid health professionals in becoming vigilant in the detection and reporting of ADRs and other drug induced problems.
- Anticipate the various combinations by which ADRs can be caused
- Considering drug-drug interactions and drug-food interactions which may responsible for ADRs
4. Functions of Pharmacovigilance
From early development through post-marketing, we need to master safety information to make critical decisions — and that requires more coordination than traditional safety systems provide. The best approach is one that integrates safety across both time and function. This lifecycle approach keeps precisely informed about product’s safety profile as it evolves — so we can not only meet regulatory and pharmacovigilance requirements, but also achieve our commercial goals2, 26
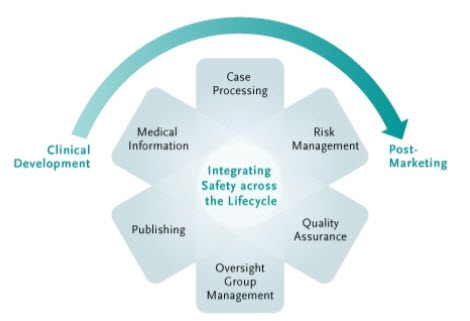
Fig.1: Different functions of Pharmacovigilance
With Quintiles, we’ll have one point of contact and access to a global array of safety and risk management professionals to help us:
- Monitor safety and manage case reports
- Mitigate risk earlier in development
- Manage and maximize your product's risk-benefit profile
- Improve the market value of your product with proactive planning and robust risk management.
5. Adverse Drug Reactions
The World Health Organization (WHO) defines an adverse drug reaction (ADR) as “Any response to a drug which is noxious and unintended, and which occurs at doses normally used in man for prophylaxis, diagnosis or therapy of disease or for modification of the physiological function”. This definition excludes overdose (either accidental or intentional), drug abuse, and treatment failure and drug administration errors. The terms “Adverse Drug Reaction” and “Adverse Drug Event” are not synonymous27.
Adverse Event
The WHO definition of an adverse event is “Any untoward medical occurrence that may present during treatment with a pharmaceutical product but which does not necessarily have a causal relationship with this treatment”. As soon as someone suspects a casual relationship between the untoward occurrence and an administered medicine, the event is turn into a suspected adverse drug reaction.
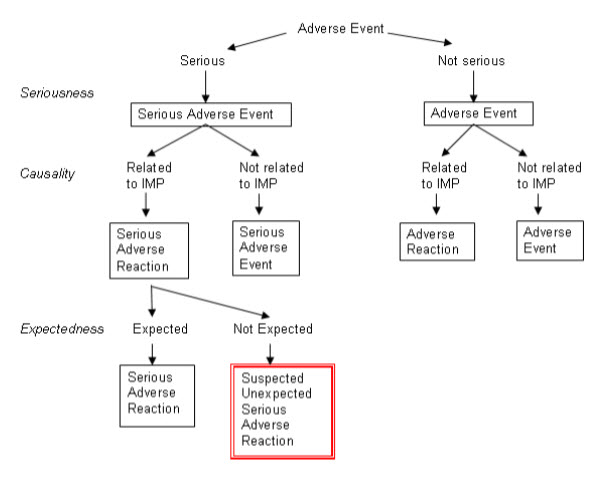
Fig.2: Adverse drug reactions flow chart
Side Effect
Any unintended effect of a pharmaceutical product occurring at doses normally used in humans, which is related to the pharmacological properties of the drug.
Unexpected adverse Reaction
An ‘Unexpected Adverse Reaction’ means an adverse reaction, the nature, severity or outcome of which is not consistent with the Summary of Product Characteristics.
Serious Adverse Reaction
A ‘Serious Adverse Reaction’ means an adverse reaction which results in death, is life threatening, requires inpatient hospitalization or prolongation of existing hospitalization, results in persistent or significant disability or incapacity or is a congenital anomaly/birth defect.
Abuse of Medicinal Products
Abuse of medicinal products’ means the persistent or sporadic, intentional excessive use of medicinal products which is accompanied by harmful physical or psychological effects.
Classification
Traditionally, ADRs are classified into two categories
- Type A
- Type B
Type A ADRs
Type A (Augmented) reactions are usually the exacerbation of pharmacological effects of a drug and thus are dose dependent. An example is insulin induced hypoglycemia. These reactions are usually predictable due to the known pharmacology of a drug and thus preventable. The incident of occurrence of type A reactions is high in any society; they are responsible for considerable morbidity. The morbidity rate is relatively low since most type A reactions will disappear by reduction of the dose or by discontinuation of drug.
Type B ADRs
Type B (Bizarre) reactions are hypersensitivity reactions and are not dose dependent. An example of penicillin induced hypersensitivity reaction. These reactions are often not predictable and preventable (unless the patient has a known history of this type of reaction). This type of reaction is rare but often serious with a high mortality rate.
The basic difference between Type A ADRs and Type B ADRs describes
Recently different newer classifications have been proposed. One of these classifications include
Type A (augmented) Type B (Bizarre)
Type C (Continuous) Type D (Delayed effect)
Type E (Rebound effect) Type F (Failure of therapy)
Wills and Brown have proposed another classification on the basis of mechanism into eight new categories which includes
Type A (augmented) Type B (Bizarre)
Type C (Chemical) Type D (Delivery)
Type E (Exit) Type F (Familial)
Type G (Genotoxicity) Type H (Hypersensitivity) Type U (Unclassified)
Table.1: Differentiation between type ADRs and type B ADRS
|
S. No. |
Type A ADRs |
Type B ADRs |
|
1. |
These type of reactions are dose dependents. |
There is no simple relationship with the dose of the drugs. |
|
2. |
They are predictable to known pharmacology. |
They are not predictable. |
|
3. |
Genetic factors may be important in case of these reactions. |
These reactions are dependent on host factors |
|
4. |
These reactions are very common in frequency. |
These reactions are uncommon in frequency. |
|
5. |
They are variable but usually mild. |
They are also variable but more severe than type A reactions. |
|
6. |
They show high morbidity. |
They also show high morbidity. |
|
7. |
They possess low mortality. |
They posses high mortality. |
|
8. |
They calculated as 80% overall proportion of ADRs. |
They calculated as 20% of overall ADRS. |
|
9. |
These reactions are first detected in phase I & III of clinical trials. |
These reactions are detected in Phase IV and occasionally in phase III. |
|
10. |
These reactions are usually because of parent drug or stable metabolite. |
These reactions may be because of parent drug or stable metabolites. |
Factors which predisposing ADRs
There are many factors that can predispose to the occurrence of adverse drug reactions in a patient. Patients who have one or more following predisposing factors are at high risk of developing ADRs.
* Polypharmacy
* Multiple and intercurrent disease
* Age
* Drug characteristics
* Gender
* Race and genetics
Polypharmacy
Patients with multiple drug therapy are more prone to develop as adverse drug reaction either due to alteration of drug effect through an interaction mechanism or by synergistic effect. The amount of risk associated with multiple drug therapy increases in direct proportion to the number of drug administered4.
Multiple and intercurrent disease
Patient with multiple diseases are at an increased risk of developing an ADR due to multiple drug used for their multiple diseases. Similarly, patients with impaired hepatic or renal status are also at a high risk of developing an ADR to drugs which are eliminated by these organs. For example a patient with decreased renal function who is treated with amino glycosides is at an increased risk of developing nephrotoxicity unless appropriate dosage adjustment is made6,9.
Age
Elderly and pediatric patients are more vulnerable to develop ADRs. Elderly patients are more susceptible to ADRs due to the physiological changes (pharmacokinetics and pharmacodynamic) which accompany aging, and also because they are often taking many drugs for chronic and multiple diseases. Nitrates or angiotensin converting enzyme inhibitor induced postural hypotension in an elderly patient is an example of this kind, where the reaction may be exacerbated by age related impaired baroreceptor responses to a change in posture. Pediatric patient may develop serious adverse drug reactions to some drugs since all children, especially neonates, differ in their drug handling capacity compared to adults. An example of such a serious reaction is the Gray Baby Syndrome with chloramphenicol.
Drug characteristics
Some drugs are highly toxic in nature and patients who are treated with these drugs are at an increased risk of ADRs. For examples, nausea and vomiting is a common adverse drug reaction seen in patients treated with anticancer drugs. Also, patients who are treated with drugs which have narrow therapeutic index such as digoxin and gentamicin are more susceptible to develop ADRs, as a slight increase in the serum drug concentration of these drugs may result in drug toxicity.
Gender
It has been reported that women are more susceptible to develop ADRs, for unknown reasons. Chloramphenicol induced aplastic anemia and phenylbutazone induced agranulocytosis in twice and trice as common, respectively in women patients.
Race and genetics
It is evident that ADRs are more common in genetically predisposed individual. For example, patients who are genetically deficient of Glucose-6-Phosphate Dehydrogenase (G6PD) enzyme are at higher risk of developing hemolysis due to primaquine than those who are not, race and genetic polymorphism may account for alterations in handling of drugs and their end organ effects.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Mechanism of type A ADRs
A drug suspected to have caused an adverse drug reaction in one patient may not necessarily cause the similar adverse drug reaction in another patient. This is due to inter individual variability, which may predispose an individual to an ADR. Any type A reaction, which occurs in an individual, may be attributed to any one or more of the following mechanism27.
* Pharmaceutical cause
* Pharmacokinetic cause
* Pharmacodynamic cause
Pharmaceutical cause
The possible pharmaceutical causes which might be attributed to occurrence of type A adverse drug reaction includes changes in the drug quantity present in a particular product, and also changes in its drug release properties. For examples, there are several reported reactions of gastrointestinal bleeding and hemorrhage due to a rate-controlled preparation of indomethacin. This may be due to irritant effects of high concentrations of indomethacin on a localized area of gastrointestinal mucosa. Another example is the corrosive effects on the esophagus caused are certain doxycycline salts. By changing to another salt, the risk may be reduced dramatically. To avoid such reactions, the drug regulatory authorities have laid down stringent requirements for marketed drug products28.
Pharmacokinetic causes
Alterations in the absorption, distribution, metabolism and elimination of drugs may alter drug effects by changing the concentration of drug present at site of action. The change in the drug effect due to alterations in pharmacokinetics parameters may be experienced as either therapeutic failure or toxicity.
Absorption: Alterations in the rate and extent of drug absorption may result in adverse drug effects. The plasma concentration of drug is partly determined by the rate at which the drug is absorbed after ingestion or injection. The plasma concentration of an orally administered drug in turn depends greatly on the gastric emptying rate. Similarly, the extent of drug absorption (the total amount of drug reaching the general circulation) also plays an important role in altered in response. Following oral administration, many factors may influence the extent of drug absorption including drug formulation, gastrointestinal motility, and first pass metabolism, concomitant administration of other drugs and the absorptive capacity of gastrointestinal mucosa. Any absorption may result in either therapeutic failure or toxicity.
Distribution: There are several factors, which determine in the extent of distribution of a drug including regional blood flow, membrane permeability and protein and tissue binding. Changes in drug distribution may predispose to adverse drug reactions, where the clinical significance of such mechanisms is yet to be proved.
Metabolism: The drug handling capacity of an individual can greatly affects the drug effect. In an individual who has a reduced metabolic rate, accumulation of drug in the body may be higher, leading to increased risk of adverse drug reactions especially type A reaction, while therapeutic failure may occur in an individual who has an enhanced metabolic rate. These changes are due to inter-individual variations in drug metabolizing capacity, which in turn is greatly influenced by genetic, environmental and other factors. For example, slow acetylators are highly prone to develop type A reaction to drugs which are metabolized by acetylation, such as isoniazid, dapsone, hydralazine and procainamide. Also differences in then rate of oxidation by cytochrome P450 system are of clinical importance.
Elimination: The major routes of excretion for many drugs are the kidneys (excretion through urine) and liver (yields metabolites which are then excreted by the kidneys). One of the most important causes of type A adverse drug reaction is a change in the drug elimination rate. Drug accumulation due to reduced elimination may predispose to adverse drug reactions as a result of increased drug concentration in plasma and tissue. Conversely, reduced concentration of drug in plasma and tissue due to enhanced drug elimination may lead to therapeutic failure.
Pharmacodynamic causes
Increased sensitivity of target tissues or organs may predispose a person to adverse drug reactions. The reasons why different individuals react differently to drugs are still not clear, evidence is accumulating to suggest that target tissue or organ sensitivity is influenced by the drug receptors themselves, by homeostatic mechanisms and by disease.
Drug receptors: Most drugs elicit their response by combining with receptors. These receptors are either protein molecules or enzymes. The amount and sensitivity of receptors of an individual may differ from another individual. Some individuals may have fewer specific drug receptors while others may have a higher number of less active receptors. This inter variability between different individuals can greatly affects the drug effect, when the drug acts through these specific receptors.
Homeostatic mechanisms: Many physiological factors may determine the extent of a drug’s effect in an individual as drug affects occur within the environment of the body’s physiological mechanisms. For examples, intravenous atropine produces a variable increase in heart rate and some individuals develop tachycardia of 160 beats per minutes at a dose which is almost ineffective in others. The magnitude of the observed effects is dependent on the balance between parasympathetic and sympathetic cardiac tone, which appears to be under genetic control.
Disease: The pharmacological effects of a drug which are not apparent in a healthy individual may be unmasked by intercurrent diseases. An example is an asthmatic patient who develops bronchoconstriction while taking non-selective beta-blockers such as propranolol.
Mechanism of type B ADRs
The type B adverse drug reactions are aberrant in terms of the normal pharmacology of the drug and they are a heterogeneous group of unpredictable adverse effects. By definition, type B reactions are unrelated to the pharmacology. The major sources for type B reactions includes decomposition of the active ingredient, the effects of the non-drug excipients (additives, preservatives, coloring and solubilising agents) and synthetic byproducts of active constituents. In the majority of cases the use of decomposed drug products may result in therapeutic failure. In some instances, though not all, the decomposed product may be highly toxic and lethal. Deaths have been reported due to decomposition of paraldehyde and its subsequent oxidation to acetic acid. There is a clear recognition of adverse drug reaction caused by excipients. Many additives including propylene glycol and carboxymethylcellulose may cause hypersensitivity reactions. The eosinophilia myalgia syndrome associated with L-tryptophan may be related to the use of preparations containing a contaminant, although a genetic factor may also be involved1,4, 29.
The metabolism of a drug to unusual reactive metabolites may give rise to type B reactions either by a direct or though an immune mediated mechanism. Examples of such reactions include phenacetin-induced methemoglobinemia and carbamazepine induced hypersensitivity reactions. Individuals whose specific bio-inactivation pathways are either more active or less active and with immunological characteristics which render them highly responsive to immunogens and haptogens are more susceptible. The reasons for the occurrence of type B reactions in a particulars individual are not clear30.
The importance of reporting ADR
Not all the hazards can be known before a medicine is marketed. Patient’s consumers and indeed some healthcare professionals may have expectations that medicinal products available are “safe” and may be surprised when regulatory action is taken to restrict the use or withdraw medicines as a result of previously unrecognized safety concerns. Information collected during the pre-marketing phase of medicinal products is inevitably incomplete with regard to complete safety profile of a product because
* Animal testing is insufficiently predictive of human safety
* Data from clinical trials is limited by their size and duration
* Patients in clinical trials are selected and the conditions of use differ from those in clinical practice
* Informations are rare but serious adverse reactions, chronic toxicity, and use in special groups such as pediatrics, geriatrics and in pregnancy) or drug interactions is often incomplete or not available and will only manifest after drug is released, may be after several years.
The effects of ADRs
All medicines have the potential to cause ADRs. Prevention of ADRs helps in order to minimize the harmful effects resulting from ADRS, harmful effects includes
* ADRs are estimated that 2-6% of all admissions to hospitals are due to ADRs
* ADRs contribute to an increased attendance to primary care and may complicate hospital in-patient stay in as many as 10% to 20% of patients
* ADRs may be responsible for deaths (Possibly as high as the 4th commonest cause of death)
* ADRs may increase the length of hospital stay and increase the cost of patient care
* ADRs may adversely affect patient quality of life and may cause patients to lose confidence in doctors
* ADRs may also mimic disease and result in unnecessary investigations and/or delay in treatment.
* ADRs are a major economic burden.
* Occurrence of toxicity in a minority of patients might preclude use of the drug in the majority of patients, if risk factors cannot be identified and appropriate regulatory measures implemented.
Pre marketing studies
The safeties of new medicines are tested in animal models. A great deal of risk information may be obtained from such tests, for examples the level of acute toxicity, which organs will be affected in case of toxicity and dose dependency of such tissue injuries. Specific animal tests for carcinogenicity, teratogenicity and mutogenicity are also available. Animals can only serve as approximately models for humans. We do not have sufficient knowledge to extrapolate information collected from animal studies directly into risks in humans. The predictive value of the different animals tests do not reveal particularly worrying results, safety tests precede onto testing in humans in clinical trial programs.
Clinical trials are carried out in three different phases prior to the submission of a marketing authorization application, with a stepwise increase in the number of individuals being exposed. Prior to the general release of a new product, not more than 4000 individuals have normally been exposed to the new drug. This implies that clinical trials, normally, only have the power to identify adverse reactions of a frequency greater that 0.5-1.0 percent. Clinical trial programs are designed to maximize the chance of demonstrating a therapeutic effect in relation to a control group. Children and the elderly are normally actively excluded from the studies. Once the drug is used in clinical practice, children and elderly, and patients with much more complicated diseases situations can be treated. For cost reasons clinical trials often have a very short duration, which means they cannot generate information about long term adverse effects31-34.
The consequence of the above is that at the time of general marketing of a new medicine, only the most common, dose related (type A) adverse reactions will be known.
Post-marketing surveillance
The most sensitive, powerful and cost effective system for identification of unknown drug related risks are spontaneous adverse reaction reporting. Every healthcare practitioner should see it as a part if his/her professional duty unexpectedly causing a risk situation for a patient under his/her cares. Pharmacovigilance should not be limited to the reporting of classical adverse effects. It should also be concerned with identification of product defects, unexpected insufficient therapeutic effects, intoxications and misuse abuse situation.
An important outcome of spontaneous adverse reaction reporting programs is the creation of signals of previously unknown or insufficiently documented problems. To verify the hypothesis of a causal link between drug exposure and an adverse outcome, it may be necessary to employ epidemiological techniques. With such techniques it is also possible to quantify the risk which is not possible through spontaneous reporting.
The two epidemiological methods that are most commonly used are cohort studies and case-control studies. In Cohort studies, patient exposed to a particular drug are followed up regularly and adverse reactions in them are compared to a matched control population. Case-control studies involve studying patients who have been affected by an adverse reactions and linking it with drug used prior to the reaction. Case control studies are particularly useful for the study of rare adverse reactions5.
In the hospital setup, healthcare professionals should be very vigilant in detecting ADRs. The possibility of an ADR should always be considered during differential diagnosis. ADRs may be detected during wards rounds with the medical team or during review of the patient’s chart. Patient counseling, medication history interview and communicating with other healthcare professionals may provide additional clues which may be useful in the detection of ADRs7.
To assist the detection of ADRs, healthcare professionals should closely monitor patients who are at risk. These includes
* Patients with renal or hepatic impairment
* Patient taking drugs which have potential to cause ADRs
* Patients who have had previous allergic reactions
* Patients taking multiple drugs
The first step in the detection of ADRs is collection of data. The data to be collected includes patient’s demographic data, presenting complaints, past medication history, drug therapy details including over the counter drugs, current medications and medication on admission, and lab data such as hematological, renal and hepatic function test. Details of the suspected adverse reaction such as time of onset and duration of reaction, nature and severity of reaction, details of the suspected drugs including dose, frequency, time of administration, duration of treatment, plasma concentration of drug, previous report on reported reaction, data on any other causes including risk factors and predisposing factors are useful11.
All the above stated relevant data can be obtained from the following sources of information
* Patient’s case notes and treatment chart
* Patient interview
* Laboratory data sources
Communication with other healthcare professionals
6. Method of Pharmacovigilance
a. Passive Surveillance
Spontaneous Reports
A spontaneous report is an unsolicited communication by health care professionals or consumers to a company, regulatory authority or other organization (e.g., WHO, Regional Centers, Poison Control Centre) that describes one or more adverse drug reactions in a patient who was given one or more medicinal products and that does not derive from a study or any organized data collection scheme.
Spontaneous reports play a major role in the identification of safety signals once a drug is marketed. In many instances, a company can be alerted to rare adverse events that were not detected in earlier clinical trials or other pre-marketing studies1-5.
Spontaneous reporting systems; yellow card scheme
The yellow card scheme was established in 1964 as a result of the thalidomide tragedy. Since then, the system has become one of the major international pharmacovigilance resources. The yellow card scheme is run jointly by the MCA (the regulatory agency) and the CSM (an expert advisorycommittee to the MCA).Since 1991, the yellow card scheme has been enhanced by a new computer system, the ADROIT (Adverse Drug Reaction Online Information Tracking) system. ADROIT is different from other databases. Not only does it store the details of the report, but also the image of the yellow card in the optical system. Multiple users can view any yellow card on screen at the same time. The MCA receives approximately 20,000 yellow cards each year. The reports are prioritized so that serious adverse drug reactions receive early attention. The yellow cards are classified into seven priorities by a member of the scientific staff according to the drugs and the nature of the ADR. Priority 1 reports receive the earliest attention
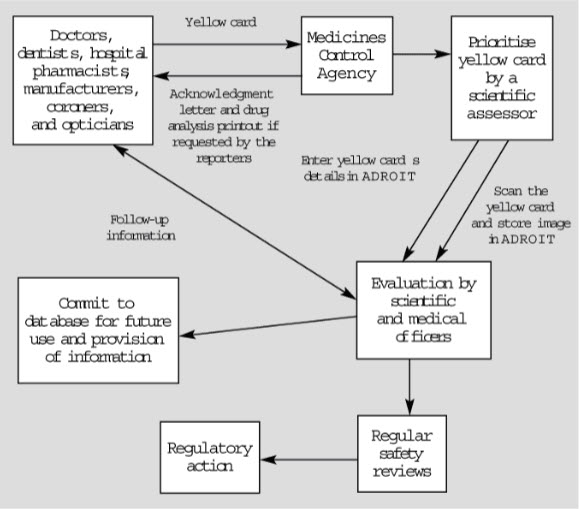
Fig. 3: Adverse drug reaction online information tracking and yellow card system
Sources of data
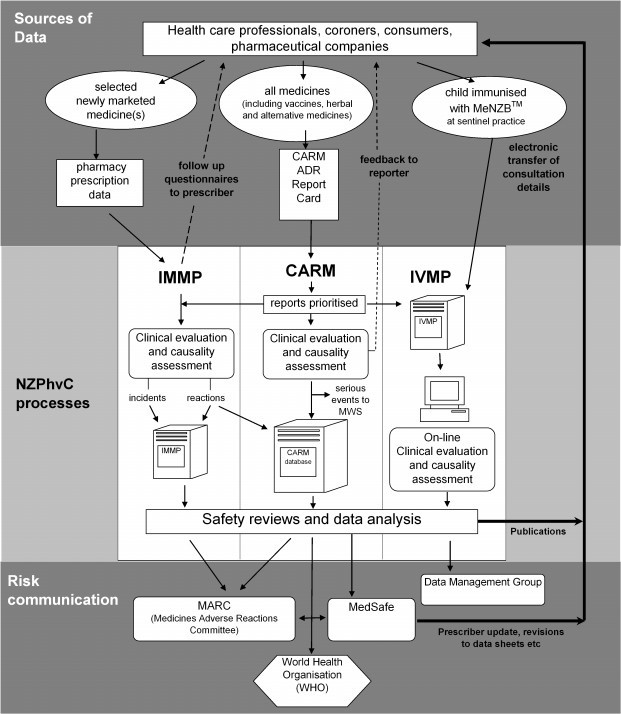
Fig. 4: Different sources of data for Pharmacovigilance
Systematic Methods for the Evaluation of Spontaneous Reports
More recently, systematic methods for the detection of safety signals from spontaneous reports have been used. These methods include the calculation of the proportional reporting ratio, as well as the use of Bayesian and other techniques for signal detection. Data mining techniques have also been used to examine drug-drug interactions. Data mining techniques should always be used in conjunction with, and not in place of, analyses of single case reports. Data mining techniques facilitate the evaluation of spontaneous reports by using statistical methods to detect potential signals for further evaluation. This tool does not quantify the magnitude of risk, and caution should be exercised when comparing drugs. Further, when using data mining techniques, consideration should be given to the threshold established for detecting signals; since this will have implications for the sensitivity and specificity of the method (a high threshold is associated with high specificity and low sensitivity). Confounding factors that influence spontaneous adverse event reporting are not removed by data mining. Results of data mining should be interpreted with the knowledge of the weaknesses of the spontaneous reporting system and, more specifically, the large differences in the ADR reporting rate among different drugs and the many potential biases inherent in spontaneous reporting. All signals should be evaluated recognizing the possibility of false positives. In addition, the absence of a signal does not mean that a problem does not exist.
Case Series
Series of case reports can provide evidence of an association between a drug and an adverse event, but they are generally more useful for generating hypotheses than for verifying an association between drug exposure and outcome. There are certain distinct adverse events known to be associated more frequently with drug therapy, such as anaphylaxis, aplastic anemia, toxic epidermal necrolysis and Stevens - Johnson syndrome. Therefore, when events such as these are spontaneously reported, sponsors should place more emphasis on these reports for detailed and rapid follow-up.
b. Stimulated Reporting
Several methods have been used to encourage and facilitate reporting by health professionals in specific situations (e.g. in-hospital settings) for new products or for limited time periods. Such methods include on-line reporting of adverse events and systematic stimulation of reporting of adverse events based on a pre-designed method. Although these methods have been shown to improve reporting, they are not devoid of the limitations of passive surveillance, especially selective reporting and incomplete information.
During the early post-marketing phase, companies might actively provide health professionals with safety information and at the same time encourage cautious use of new products and the submission of spontaneous reports when an adverse event is identified. A plan can be developed before the product is launched (e.g., through site visits by company representatives, by direct mailings or faxes, etc.). Stimulated adverse event reporting in the early post-marketing phase can lead companies to notify healthcare professionals of new therapies and provide safety information early in use by the general population (e.g., Early Post-marketing Phase Vigilance, EPPV in Japan). This should be regarded as a form of spontaneous event reporting, and thus data obtained from stimulated reporting cannot be used to generate accurate incidence rates, but reporting rates can be estimated.
c. Active Surveillance
Active surveillance, in contrast to passive surveillance, seeks to ascertain completely the number of adverse events via a continuous pre-organised process. An example of active surveillance is the follow-up of patients treated with a particular drug through a risk management program. Patients who fill a prescription for this drug may be asked to complete a brief survey form and give permission for later contact. In general, it is more feasible to get comprehensive data on individual adverse event reports through an active surveillance system than through a passive reporting system.
Sentinel Sites
Active surveillance can be achieved by reviewing medical records or interviewing patients and/or physicians in a sample of sentinel sites to ensure complete and accurate data on reported adverse events from these sites. The selected sites can provide information, such as data from specific patient subgroups that would not be available in a passive spontaneous reporting system. Further, information on the use of a drug, such as abuse, can be targeted at selected sentinel sites. Some of the major weaknesses of sentinel sites are problems with selection bias, small numbers of patients, and increased costs. Active surveillance with sentinel sites is most efficient for those drugs used mainly in institutional settings such as hospitals, nursing homes, haemodialysis centers, etc. Institutional settings can have a greater frequency of use for certain drug products and can provide an infrastructure for dedicated reporting. In addition, automatic detection of abnormal laboratory values from computerized laboratory reports in certain clinical settings can provide an efficient active surveillance system. Intensive monitoring of sentinel sites can also be helpful in identifying risks among patients taking orphan drugs.
Drug Event Monitoring
Drug event monitoring is a method of active pharmacovigilance surveillance. In drug event monitoring, patients might be identified from electronic prescription data or automated health insurance claims. A follow-up questionnaire can then be sent to each prescribing physician or patient at pre-specified intervals to obtain outcome information. Information on patient demographics, indication for treatment, duration of therapy (including start dates), dosage, clinical events, and reasons for discontinuation can be included in the questionnaire. Limitations of drug event monitoring can include poor physician and patient response rates and the unfocused nature of data collection, which can obscure important signals32.
Registries
A registry is a list of patients presenting with the same characteristic(s). This characteristic can be a disease (disease registry) or a specific exposure (drug registry). Both types of registries, which only differ by the type of patient data of interest, can collect a battery of information using standardised questionnaires in a prospective fashion. Disease registries, such as registries for blood dyscrasias, severe cutaneous reactions, or congenital malformations can help collect data on drug exposure and other factors associated with a clinical condition. A disease registry might also be used as a base for a case-control study comparing the drug exposure of cases identified from the registry and controls selected from either patients with another condition within the registry, or patients outside the registry.
Exposure (drug) registries address populations exposed to drugs of interest (e.g., registry of rheumatoid arthritis patients exposed to biological therapies) to determine if a drug has a special impact on this group of patients. Some exposure (drug) registries address drug exposures in specific populations, such as pregnant women. Patients can be followed over time and included in a cohort study to collect data on adverse events using standardised questionnaires. Single cohort studies can measure incidence, but, without a comparison group, cannot provide proof of association. However, they can be useful for signal amplification, particularly for rare outcomes. This type of registry can be very valuable when examining the safety of an orphan drug indicated for a specific condition.
d. Comparative Observational Studies
Traditional epidemiologic methods are a key component in the evaluation of adverse events. There are a number of observational study designs that are useful in validating signals from spontaneous reports or case series. Major types of these designs are cross-sectional studies, case-control studies, and cohort studies (both retrospective and prospective).
Cross-Sectional Study (Survey)
Data collected on a population of patients at a single point in time (or interval of time) regardless of exposure or disease status constitute a cross-sectional study. These types of studies are primarily used to gather data for surveys or for ecological analyses. The major drawback of cross-sectional studies is that the temporal relationship between exposure and outcome cannot be directly addressed. These studies are best used to examine the prevalence of a disease at one time point or to examine trends over time, when data for serial time points can be captured. These studies can also be used to examine the crude association between exposure and outcome in ecologic analyses. Cross-sectional studies are best utilized when exposures do not change over time.
Case-Control Study
In a case-control study, cases of disease (or events) are identified. Controls, or patients without the disease or event of interest, are then selected from the source population that gave rise to the cases. The controls should be selected in such a way that the prevalence of exposure among the controls represents the prevalence of exposure in the source population. The exposure status of the two groups is then compared using the odds ratio, which is an estimate of the relative risk of disease in the two groups. Patients can be identified from an existing database or using data collected specifically for the purpose of the study of interest. If safety information is sought for special populations, the cases and controls can be stratified according to the population of interest (the elderly, children, pregnant women, etc.). For rare adverse events, existing large population-based databases are a useful and efficient means of providing needed drug exposure and medical outcome data in a relatively short period of time. Case-control studies are particularly useful when the goal is to investigate whether there is an association between a drug (or drugs) and one specific rare adverse event, as well as to identify risk factors for adverse events. Risk factors can include conditions such as renal and hepatic dysfunction, which might modify the relationship between the drug exposure and the adverse event. Under specific conditions, a case-control study can provide the absolute incidence rate of the event.
If all cases of interest (or a well-defined fraction of cases) in the catchment area are captured and the fraction of controls from the source population is known, an incidence rate can be calculated.
e. Targeted Clinical Investigations
When significant risks are identified from pre-approval clinical trials, further clinical studies might be called for to evaluate the mechanism of action for the adverse reaction. In some instances, pharmacodynamic and pharmacokinetic studies might be conducted to determine whether a particular dosing instruction can put patients at an increased risk of adverse events. Genetic testing can also provide clues about which group of patients might be at an increased risk of adverse reactions. Furthermore, based on the pharmacological properties and the expected use of the drug in general practice, conducting specific studies to investigate potential drug-drug interactions and food-drug interactions might be called for. These studies can include population pharmacokinetic studies and drug concentration monitoring in patients and normal volunteers.
Sometimes, potential risks or unforeseen benefits in special populations might be identified from pre-approval clinical trials, but cannot be fully quantified due to small sample sizes or the exclusion of subpopulations of patients from these clinical studies. These populations might include the elderly, children, or patients with renal or hepatic disorder. Children, the elderly, and patients with co-morbid conditions might metabolise drugs differently than patients typically enrolled in clinical trials. Further clinical trials might be used to determine and to quantify the magnitude of the risk (or benefit) in such populations.
f. Descriptive Studies
Descriptive studies are an important component of phannacovigilance, although not for the detection or verification of adverse events associated with drug exposures. These studies are primarily used to obtain the background rate of outcome events and/or establish the prevalence of the use of drugs in specified populations.
Natural History of Disease
The science of epidemiology originally focused on the natural history of disease, including the characteristics of diseased patients and the distribution of disease in selected populations, as well as estimating the incidence and prevalence of potential outcomes of interest. These outcomes of interest now include a description of disease treatment patterns and adverse events. Studies that examine specific aspects of adverse events, such as the background incidence rate of or risk factors for the adverse event of interest can be used to assist in putting spontaneous reports into perspective33.
Uppsala Monitoring Centre26
The principal function of the Uppsala Monitoring Centre is to manage the international database of ADR reports received from National Centres. In 2002 this database held nearly three million case reports. The majority of national contributing centres have easy electronic access to these. The UMC has established standardized reporting by all National Centres and has facilitated communication between countries to promote rapid identification of signals.
A sophisticated Bayesian confidence propagation neural network (BCPNN) programme was created in 1998, which partly automates the signal detection system, and provides earlier alert signals than previous methods.
The effectiveness of this system depends on:
• The size of the database
• The quality of the reports received from the contributing centres
• The timeliness of such reporting
• An active and reliable reporting culture within participating countries.
An international advisory panel of clinical experts determines the validity and clinical importance of the signals generated.
In recent years the UMC has expanded its role as a communications and training centre and clearing-house for information on drug safety. Through
1. Mail discussion groups
2. Website development
3. newsletters
4. Annual national centre meetings
The UMC team, in collaboration with the WHO, facilitates and encourages the international collaboration, which was identified in 1972 as being vital for the success of pharmacovigilance.
The terminologies developed within the WHO programme for coding adverse reactions and medicines have been widely adopted by National Centres, manufacturers and drug regulators. In recent years, the introduction of a new terminology known as MedDRA (Medical Dictionary for Drug Regulatory Activities) has replaced the World Health Organization Adverse Reaction Terminology (WHO-ART) in developed countries. WHO-ART remains the mainstay of communicating adverse reactions in most developing countries within the International Programme34.
Another project at the UMC is the creation of an ADR monitoring system for herbal and traditional medicines.
While the UMC has achieved much in improving the activities, support and recognition of individual National Centres, much more could still be done in providing training and encouraging expertise at a national level. There needs to be better consultation and communication between developed and developing countries when discussions on international harmonization of pharmacovigilance issues are taking place.
Clinical trial regulation
In recent years there has been a substantial increase in the number of clinical trials in developed and developing countries. Clinical trials in the United States of America alone nearly doubled between 1990 and 1998. With sequencing of the human genome, clinical research in potential new drug therapies is likely to increase even further.
There is also a growing alliance between academia and the pharmaceutical and biotechnology industries. This has given rise to serious and widespread concern over ethical and scientific issues such as:
1. The potential for conflict of interest.
2. Unethical patient recruitment practices.
3. Inadequacy of informed consent.
4. Lack of capacity to ensure on-going monitoring of clinical trials and adherence to principles of sound and ethical clinical practice.
5. Poor reporting and management of adverse events.
For drug regulators, the changing trends over recent years in the conduct of clinical trials present special and urgent challenges, particularly in ensuring that the rights and health of patients and their communities are protected. In their approval of clinical trials, regulatory bodies look at safety and efficacy of new products under investigation. They must also pay attention to the general standards of care and safety of study subjects, in conjunction with the appropriate institutional review boards (IRBs).
Medicines those are required for diseases such as tuberculosis, malaria, HIV/AIDS and meningococcus A meningitis, and those which may have a questionable or uncertain effectiveness - safety profile, require careful surveillance when first introduced on a large scale into communities.
The increasing complexity of clinical trials presents further challenges to regulators. Study designs often require large cohorts of participants. In many instances trials are carried out at various sites in several countries. Local ethics committees and drug regulators are not always aware of patients’ and investigators’ experiences at other international sites. Clinical trials are increasingly contracted to clinical research organizations and patient recruitment agencies, which act as intermediaries between the sponsors of the study, the investigators and the patients.
Responsibility for ensuring proper conduct of the clinical trial may, in such circumstances, be divided between the parties. Information requested by ethics committees and regulators may be difficult to obtain in a short time. Regulators and ethics committees do not always have the capacity to carry out these functions effectively. This may have serious implications for the safety of patients8, 35.
Safety monitoring during clinical trials is now recognized as one of the major concerns for new drug development. This is currently being addressed by a CIOMS working group. Three main topics are being addressed:
1) The collection of adverse experience information
2) Assessment/monitoring of clinical data
3) Reporting/communication of clinical data.
A standardized reporting system for safety concerns arising during clinical trials might serve as a helpful tool for regulatory agencies, and for ethics committees (institutional review boards), provided there were full exchange of information between them and the investigators and sponsors. Expedited electronic submission of safety reports in ICH countries has facilitated the reporting process to some extent; however, routine review of safety information requires considerable resources, expertise, support and commitment from those involved.
Once research into new drugs is in the post-marketing stage (Phase IV studies) safety may be monitored to comply with the conditions of registration, particularly when there are unresolved concerns. This may lead to improved and more rapid changes in labeling or even withdrawal of a new drug from the market. Routine application of principles of good clinical practice that ensure patient safety and strict compliance with prescribed regulatory requirements would substantially improve standards of clinical trials36.
Pharmacovigilance and the national drug regulatory authority
The limitations of pre-marketing drug safety data are well-recognized. They are aggravated by increasing pressure on drug regulators from the pharmaceutical industry to shorten the review time for new medicines. Registration approval of a new drug is likely to be followed by robust marketing and rapid exposures of thousands even millions of patients to it. The implications for drug safety of this evolving situation need to be addressed.
For example, it is necessary that in the proper conduct of pharmacovigilance there should be access to the information on which the original determination of risk and harm was made. Pre-registration files, including the advice and opinions of the original evaluators of the data, are required if a balanced and clinically relevant decision is to be made. Beyond response to events and media reporting, active enquiry and detailed clinical investigation are the essential tools of this work37.
Herbal and Traditional Medicines
The use of herbal and traditional medicines raises concerns in relation to their safety. There is wide misconception that ‘natural’ means ‘safe’. There is the common belief that long use of a medicine, based on tradition, assures both its efficacy and safety. There are examples of traditional and herbal medicines being adulterated or contaminated with allopathic medicines, chemicals such as corticosteroids, non-steroidal anti-inflammatory agents and heavy metals. Many traditional medicines are manufactured for global use and they have moved beyond the traditional and cultural framework for which they were originally intended. Self-medication further aggravates the risk to patients. When traditional and herbal medicines are used in conjunction with other medicines there is the potential of serious adverse drug interactions.
These disparities in regulation between countries have serious implications for international access to and distribution of such products. For instance, in one country a herbal product may be obtainable only on prescription and from an authorized pharmacy, whereas in another country, it may be obtainable from a health food shop, or even, as has become common practice, by mail order or Internet.
For all these reasons, inclusion of herbal and traditional medicines in national pharmacovigilance programmes has become important and inevitable. Healthcare providers, including traditional health practitioners, regulators, manufacturers and the public share a responsibility for their informed and safe use. The World Health Organization has produced guidelines for assessment of the safety, efficacy and quality of herbal medicines.
Vaccines and biological medicines
For several reasons, vaccines and biologicals require modified systems of safety monitoring. They are often administered to healthy children. This applies particularly to vaccines used within a national immunization programme. In many countries, those exposed to a particular vaccine represent the entire birth cohort and therefore a sizeable part of the entire population. People’s expectations of safety are high, and they are reluctant to countenance even a small risk of adverse events. Concerns regarding vaccine safety, real or imagined, may result in loss of confidence in entire vaccine programmes. This can result in poor compliance and a consequent resurgence in morbidity and mortality of vaccine-preventable disease.
The difficulties in monitoring and dealing with vaccine safety are complicated by the problems inherent in determining the causal link between an adverse event following immunization and a vaccine. For example, information on dechallenge and rechallenge is often missing, and vaccines are given to most of the country’s birth cohort at an age when coincidental disease is likely. Several vaccines are likely to be administered concurrently. The possibility of programmatic errors should never be overlooked. (A programmatic error is a medical incident that was caused by some error in the transportation, storage, handling or administration of vaccines.)
However, the responsibility of the regulatory authority is by no means limited to the safety of vaccines used in immunization programmes. Several biological products are used in specific patient populations as preventive or curative measures. The efficient regulation of these products is crucial in order to avoid potential harm to the public as a result of substandard manufacture or improper transportation and storage of imported vaccines and biologicals.
In recent years, the safety of biological products and blood products has come under public scrutiny. Concerns about the safety of medicinal products of animal origin have been raised in connection with variant Creutzfeldt-Jacob disease (vCJD), and with contamination of blood and blood products by infectious organisms such as HIV and hepatitis B. The quality of screening and sterilization procedures and appropriate selection of donors are linked to the risks of contamination. Such safety issues related to the use of plasma-derived medicinal products should fall under the aegis of pharmacovigilance programmes. For that to happen, pharmacovigilance centres would have to consider the special issues related to safety of these products. Expertise in biological products, virology and medical microbiology would be required.
New vaccines for pandemic diseases such as HIV/AIDS and malaria are in the later phases of development. Clinical trials in large patient populations are being considered for testing the efficacy and safety of these vaccines. Special ethical, legal and regulatory challenges are raised in the conduct of such clinical trials, especially the implications vaccines may have for the epidemiology of disease and the possible direct and indirect risks of harm associated with the introduction of vaccines into large communities37.
Scope of Phramacovigilance
The discipline of pharmacovigilance has developed considerably since the 1972 WHO technical report, and it remains a dynamic clinical and scientific discipline. It has been essential to meet the challenges of the increasing range and potency of medicines (including vaccines), which carry with them an inevitable and sometimes unpredictable potential for harm. The risk of harm, however, is less when medicines are used by an informed health profession and by patients who themselves understand and share responsibility for their drugs. When adverse effects and toxicity appear particularly when previously unknown in association with the medicine - it is essential that they should be analysed and communicated effectively to an audience that has the knowledge to interpret the information. This is the role of pharmacovigilance. Much has already been achieved. But more is required for the integration of the discipline into clinical practice and public policy2, 5, 8, 38.
The following is a summary of some of the serious challenges facing pharmacovigilance programmes in the next ten years, describing in brief the potential implications of such trends on the evolution of the science.
Major challenges are:
I. Globalization
The globalization of drug distribution and the increased exposure of massive populations to large volumes of medicines. These include novel chemical entities used for symptomatic relief and lifestyle modification as well as medicines used in developing countries to curb the prevalence of pandemic diseases such as
HIV / AIDS, malaria and tuberculosis. The use of medicines on such a large scale and within such a short period of time calls for a better and more efficient level of international pharmacovigilance.
II. Web-based sales and information
The Internet, in addition to its many benefits, has also facilitated the uncontrolled sale of
medicines (including herbal and traditional medicines) across national borders. Drug information in all forms and with varying levels of accuracy is distributed internationally through this medium. Such information covers: prescription drugs, unregistered medicines, highly controlled substances and traditional and herbal medicines with questionable safety, efficacy and quality. Regulatory decisions on drug safety made in distant countries are available to the international public at the same time as national drug regulatory authorities. Aggressive marketing by manufacturers and distributors through the Internet often results in excessive and, probably irrational, use of medicines. All these changes in drug use are likely to have important consequences on public health and safety.
III. Broader safety concerns
The scope of pharmacovigilance continues to broaden as the array of medicinal products grows. There is a realization that drug safety is more than the monitoring, detection and assessment of ADRs occurring under clearly defined conditions and within a specific dose range. Rather, it is closely linked to the patterns of drug use within society.
Problems resulting from:
• irrational drug use
• overdoses
• polypharmacy and interactions
• increasing use of traditional and herbal medicines with other medicines
• illegal sale of medicines and drugs of abuse over the Internet
• increasing self medication practices
• substandard medicines
• medication errors
• lack of efficacy
are all within the domain of pharmacovigilance. Current systems need to evolve in order to address this broad scope adequately.
Another aspect of broadened scope is the lack of clear boundaries between:
• blood products
• biologicals
• medical devices
• cosmetics
• food additives
• vaccines.
IV. Public health versus pharmaceutical industry economic growth
There may be shortcomings and at times conflicting interests within the pharmaceutical industry when dealing with public health concerns arising from drug safety issues. The industry needs to overcome weaknesses in safety monitoring during clinical trials and post-marketing surveillance. Manufacturers should take a more proactive approach to drug safety rather than maintaining defensive tactics. This calls for a heightened level of product stewardship and recognition of responsibility to public and environmental health.
V. Monitoring of established products
The generic sector of the pharmaceutical industry has not fully recognized its responsibility to continuously monitor the safety of its products throughout the world. There is the erroneous belief that generic drugs are inherently safe even when they interact with other medicines. The generic sector is the largest supplier of essential drugs.
VI. Developing and emerging countries
Outside the GECD countries, the pharmaceutical industry has not been committed to pharmacovigilance activities, particularly the drug safety issues involving medicines used in communities with over burdened health care systems, different patterns of drug use and different co morbid conditions. Other problems to be tackled include: irrational and potentially unsafe drug donation practices, and widespread manufacture and sale of counterfeit and substandard medicines.
VII. Attitudes and perceptions to benefit and harm
These trends have dramatically changed the way in which medicines are used by society. Healthcare providers, patients and the public have responded in different ways to these changing trends as has been described in previous chapters. Their perception of benefit and harm and the level of acceptable risk for medicines in the face of these rapid developments have not been considered in a meaningful way. The harm caused by medicines has been shown to be significant. Morbidity and mortality from drug-induced diseases are only recently being recognized as an important item on the public health agenda in developed and developing countries39.
VIII. Outcomes and Impact
Along with increased public awareness over safety of medicines, there is an increasing public gaze on the performance of the health professions, industry and regulators. Increased accountability must lead to more research into the effectiveness of pharmacovigilance and its place in improving public perception. A major focus must be to empower health practitioners and patients themselves with useful information that improves individual therapy, aids the diagnosis and management of medicine-induced disease, and generally leads to a reduction of iatrogenic diseases
Pharmacovigilance by region
Europe
The pharmacovigilance effort in Europe is coordinated by the European Medicines Agency (EMA) and conducted by the national competent authorities (NCAs). The main responsibility of the EMA is to maintain and develop the pharmacovigilance database consisting of all suspected serious adverse reactions to medicines observed in the European Community. The system is called EudraVigilance and contains separate but similar databases of human and veterinary reactions40-41.
Europe requires the individual marketing authorisation holders (drug companies), to submit all received adverse reactions in electronic form (save in exceptional circumstances). The reporting obligations of the various stakeholders are defined in the Community legislation, in particular:
Regulation (EC) No 726/2004
for human medicines, European Union Directive 2001/83/EC as amended and Directive 2001/20/EC
for veterinary medicines, Directive 2001/82/EC as amended.
Reporting can be performed with software developed for the purpose or with a web utility called EVWEB accessible through the EudraVigilance homepage. Registration for use of EVWEB is necessary.
In 2002 Heads of Medicines Agencies agreed on a mandate for an ad hoc Working Group on establishing a European risk management strategy. The Working Group considered the conduct of a high level survey of EU pharmacovigilance resources to promote the utilisation of expertise and encourage collaborative working.
Japan
In Japan, pharmacovigilance is regulated by the PMDA and MHLW.
United States
See also: Regulation of therapeutic goods in the United States
Three primary branches of pharmacovigilance in the U.S. include the FDA, the pharmaceutical manufacturers, and the academic/non-profit organizations (such as RADAR and Public Citizen).
Conclusion
Pharmacovigilance required for systematically identifying and correlating drugs and side-effects and taking corrective actions, especially for the product launching first time in India. Due to the evolution of a new Patent regime in the Indian Pharmaceutical Industry it is critical to have a system of pharmacovigilance in India The Pharmacovigilance programme has been started from 23 November 2004 at New Delhi and the programme is coordinated by the Central Drugs Standard Control Organization, sponsored by the World Health (who)organization. It is related to the surveillance of drugs once they are released for use in the community (post marketing surveillance) and relies on voluntary reporting, prescription monitoring, medical records and statistical studies in the population.
References
1. FSK Barar, Essentials of Pharmacotherapeutics,2004,S,Chand and company Publication, New Delhi, 110055,3 edition,59,60
2. Padmaja Udaykumar “Text Book of Medical Pharmacology” CBS publication, 2nd edition, 4.
3. US FDA , Guidance for Industry Good Phramacovigilance Practices and Pharmacoepidemiologic Assessment, Center for Drug Evaluation and Research and Center for Biologics Evaluation and Research, Rockville, MD, March 2005
4. S George Crruthers, Brain B Hoffman, Kenneth L Melmon, David W Nierenberg, “Melmon and Morrellis, Clinical Pharmacology, MC GRAW HILL,4th edition,1333,1334
5. Brain L Storm, “Pharmacoepidemiology”WILEY,3ed edition,22-25
6. H P Rang, M M Dale, J M Ritter, P K Moore, “Pharmacology”, CURCHILL LIVINGSTONE, 5th edition,5
7. Derek G Waller, Andrew G Renwick, Keith Hillier, “Medical Pharmacology and Therapeutics” ELSEVIER SAUNDERS, 2nd edition, 55, 56
8. National Pharmacovigilance programme, “Indian Journal of Pharmacology” Medknow Publications, volume 37,issue 6, December 2005
9. Singhal S, Chakraborty BS, (2012). Signal Detection of Docetaxel in Canadian Spontaneous Adverse Event Reports. Journal of Pharmacy Research, 5, 1-6
10. SZ Rahman & KC Singhal, Problems in pharmacovigilance of medicinal products of herbal origin and means to minimize them, Uppsala Reports, WHO Collaborating Center for ADR monitoring, Uppsala Monitoring Centre, Sweden, Issue 17 January 2002: 1-4 (Supplement).
11. Rahman, SZ; Khan RA, Gupta V, Misbah Uddin (2008). "Chapter 2: Pharmacoenvironmentology – Ahead of Pharmacovigilance". In Rahman SZ, Shahid M & Gupta A. An Introduction to Environmental Pharmacology (1st Ed.). Aligarh: Ibn Sina Academy. pp. 35–52.
12. The Importance of pharmacovigilance Safety monitoring of medicinal products. WHO Lib Catalog. 2002. Available from: http://www.apps.who.int/medicinedocs/en/d/Js4893e/ [last cited on 2012, Dec 25] .
13. Effective Communications in Pharmacovigilance. The Erice Report. International Conference on Developing Effective Communications in Pharmacovigilance, Erice, Sicily. 1997. Available from: http://www.who-umc.org/DynPage.aspx?id=22690 [last cited on 2013, Jan 20],
14. WHO Policy Perspectives on Medicines. Geneva: WHO; 2004. Geneva: World Health Organization. Looking at the Pharmacovigilance: ensuring the safe use of medicines. Available from: http://www.whqlibdoc.who.int/hq/2004/WHO_EDM_2004.8.pdf [cited on 2012, Dec 15].
15. Harmark L, van Grootheest AC. Pharmacovigilance: Methods, Recent Developments and Future Perspectives. Eur J Clin Pharmacol. 2008; 64:743–52.
16. Waller patrick, an introduction to pharmacovigilance, John Wiley and Sons Ltd. Chichester, West Sussex, UK, reprint 2010 page no.-189.
17. Gunasakaran S, Kumar Satheesh R, A Practical Guide on Pharmacovigilance for Beginners, first edition, 2010, Taramani Magalir Co-operative Press, Chennai, Tamilnadu, India, page no.-250.
18. Mann D. Ronald, Andrews B Elizabeth, Pharmacovigilance second edition, 2006, John Wiley and Sons Ltd. Chichester, West Sussex, UK, page no.-135.
19. SZ Rahman, RA Khan, V Gupta & Misbahuddin. Pharmacoenvironmentology – Ahead of Pharmacovigilance. In: Rahman SZ, Shahid M & Gupta A Eds. An Introduction to Environmental Pharmacology (ISBN # 978-81-906070-4-9). Ibn Sina Academy, Aligarh, India, 2008: 35-42
20. S Z Rahman, R A Khan, Varun Kumar, Misbahuddin, Pharmacoenvironmentology – A Component of Pharmacovigilance, Environmental Health 2007, 6:20 (24 Jul 2007)
21. Ilene Sue Ruhoy, Christian G. Daughton. Beyond the medicine cabinet: An analysis of where and why medications accumulate. Environment International 2008, Vol. 34 (8): 1157-1169
22. Lindquist M. Vigibase, the WHO Global ICSR Database System: Basic Facts. Drug Information Journal, 2008, 42:409-419.
23. http://www.who-umc.org/DynPage.aspx?id=13140&mn=1514, accessed 10 February 2013.
24. Pharmacovigilance. Mann RD, Andrews EB, eds. John Wiley & Sons Ltd, Chichester, 2002.
25. WHO guidelines on safety monitoring of herbal medicines in pharmacovigilance systems, World Health Organization, Geneva, 2004
26. SZ Rahman & KC Singhal, Problems in pharmacovigilance of medicinal products of herbal origin and means to minimize them, Uppsala Reports, WHO Collaborating Center for ADR monitoring, Uppsala Monitoring Centre, Sweden, Issue 17 January 2002: 1-4 (Supplement)
27. http://heads.medagencies.org, accessed 9 July 2012.
28. Davis S, Coulson RA, Wood SM. Adverse drug reaction reporting by hospital pharmacists: the first year. Pharm J 1999; 262:366, 67.
29. Committee on Safety of Medicines/Medicines Control Agency. Current Problems in Pharmacovigilance 1993; 19:1.
30. Roberts P. Adverse drug reactions (ADRs). A distance learning pack for community pharmacists. Manchester: Centre for Pharmacy Postgraduate Education, 1994.
31. Freeman J. They test new drugs don’t they? Drug safety: a shared responsibility. Edinburgh: Churchill Livingston, 1991:13-23.
32. Rowlands S, Savage I. GPRD: Inaccuracies corrected (letter). Pharm J 1999; 262:397.
33. Wood SM, Coulson RA. Adverse drug reactions on- line information tracking (ADROIT). Pharm Med 1993; 7:203-13.
34. . Waller PC, Coulson RA, Wood SM. Regulatory pharmacovigilance in the United Kingdom: current principles and practice. Pharmacoepidemiol Drug Safety 1996; 5:363-75.
35. . Committee on Safety of Medicines/Medicines Control Agency. Current Problems in Pharmacovigilance 1993;25:112
36. Rawson NSB, Pearce GL, Inman WHW. Prescription event monitoring: methodology and recent progress. J Clin Epidemiol 1990;43:509-22
37. Mann RD. Prescription event monitoring recent progress and future horizons. Br J Clin Pharmacol 1998;46:195-201
38. Mann RD, Wilton LV, Pearce GL, Mackay FJ, Dunn NR. Prescription event monitoring in 1996. A method of nonintervention observational cohort pharmacovigilance. Pharmacoepidemiol Drug Safety 1997;6(Suppl 3):S5-S11
39. Mackay FJ, Wilton LV, Pearce GL, Freemantle SN, Mann RD. Safety of long-term lamotrigine in epilepsy. Epilepsia 1997; 38:881-6.
40. Jick H, Jick SS, Gurewich V, Myers MW, Vasilakisss C. Risk of idiopathic cardiovascular death and nonfatal venous thromboembolism women using oral contraceptives with different progestagen components. Lancet 1995;346:1589-93
41. Patidar Dindayal, Adverse drug reaction reporting, monitoring and implementation system, Holyfamily Hospital, Mumbai, 2009; 2-26







