
About Authors:
Abhisek Shukla*, Dr. Valluru Ravi
Pharmaceutical Regulatory Affairs group, Dept. of Pharmaceutics,
JSS College of Pharmacy, JSS University,
Sri Shivarathreeshwara Nagar, Mysore-570015, Karnataka, India.
abhishekjsscp@gmail.com
Abstract
A regulatory process, by which a person/organization/sponsor/innovator gets authorization to launch a drug in the market, is known as drug approval process. Every country has its own regulatory authority, which is responsible to enforce the rules and regulations and issue the guidelines to regulate the marketing of the drugs. A drug approval process comprises of various stages: application to conduct clinical trials, conducting clinical trials, application to marketing authorization of drug and post-marketing studies. The purpose of this article is to present a concise overview of the drug approval process in Turkey and Canada.
Reference Id: PHARMATUTOR-ART-1973
Introduction:
In present time each country has different regulatory body and regulatory requirement.
The single regulatory approach for marketing authorization application (MAA) of a new drug product applicable to various countries (on the basis of single dossier) is extremely difficult. Therefore, the knowledge of exact and detailed regulatory requirements for MAA of each country should be known to establish a suitable regulatory strategy.
There are two stages involved in new drug approval process.
1. Clinical trial stage
2. Marketing authorization stage
The clinical trials stage would be conduct from phase I to phase IV. These studies are performed to ensure the efficacy, safety and optimizing the dose of drug in human beings. After the completion of clinical studies of the drug, then an application to the competent authority of the concerned country for the approval of drug for marketing is submitted. The competent authority review the application and approve the drug for marketing only if the drug is found to be safe and effective in human being or the drug have more desirable effect as compare to the adverse effect.
After the approval of new drug, government should monitor its safety due to appearance of some side effects, when it is used in larger population. The interactions with other drugs, which were not assessed in a pre-marketing research trial and its adverse effects (in particular populations), should also be monitored.
New drug approval process in Turkey:
In Turkey, the Ministry of Health, General Directorate of Pharmaceuticals and Pharmacies is the sole authority in charge of registration, marketing approval/ authorisation, pricing of pharmaceuticals, legal classification and inspection. The role of this authority is to provide for registration, marketing approval/ authorisation and pricing of pharmaceutical products, to define rules to be followed as well as to control the advertisement of pharmaceutical products, to undertake inspection of pharmaceutical products and pharmaceutical production plants in Turkey. According to the World Health Organization, Turkey's drug licensing standards closely resemble the countries of the European Union.
In Turkey, regulatory approval procedures of human medicinal products are conducted in accordance with the “Registration Regulation of Human Medicinal Products,” published for effect in Official Gazette #25705 of 19.01.2005.
The objective of the Registration Regulation of Human Medicinal Products is to set forth the principles, procedures, and policies regarding registered human medicinal products, with a view to achieving the desired efficacy and safety as well as the required quality in medicinal products for human use.
In Turkey, new drugs are granted marketing authorization after reviewing their safety, efficacy and quality. The General Directorate of Pharmaceuticals and Pharmacy (IEGM) is charged with the marketing authorization process, and is the principal national authority for approval, pricing, legal classification and inspection of drugs. The General Directorate is supported by scientific committees in conducting medical, pharmaceutical and clinical evaluations of products proposed for approval. Committees evaluate documents submitted by pharmaceutical manufacturers, and their decisions provide the basis for marketing authorization and licensure. The application is initially reviewed by the Advisory Committee for Registration of Human Medicinal Products, usually taking 3 to 4 months.
The third step in the marketing authorization process involves setting the product price, which is the responsibility of IEGM Pricing Branch. Thus, pricing is a part of the regulatory approval process. The price is set using an external reference price chart. The pricing procedure usually takes 3 to 6 months. After completion of pricing negotiations, the committee reviews the application for bioequivalence (for generic products) and bioavailability (for original products).For alignment with the European Union regulations, the Registration Regulation of Human Medicinal Products requires following the Common Technical Document (CTD) guidelines for preparing the marketing authorization application file. Accordingly, the CTD guidelines were issued by the Ministry of Health, and marketing authorization applications are accepted in CTF format since 30.12.2005.
The General Directorate of Pharmaceuticals and Pharmacy was the sole authority charged with granting marketing authorization and selling permits for, and pricing, classifying and reviewing drugs in Turkey. By “Decree Law #633 on the Organization and Mandate of the Ministry of Health and Subordinate Agencies – KHK/663,” published in Official Gazette #28103, second edition, of 02.11.2011, however, a “Medicines and Medical Devices Agency of Turkey” was established, with a private budget and having the status of a public juristic person, as a subordinate organ of the Ministry of Health, charged with overseeing and regulating matters pertinent to medicines, active ingredients and excipients used in the production of medicines, substances subject to national or international control, medical devices, in vitro diagnostic medical devices, traditional herbal medicinal products, cosmetics, homeopathic medicinal products and special-purpose dietary goods in line with Ministry policies and objectives.
According to the Registration Regulation of Human Medicinal Products, the Ministry conducts a preliminary review to evaluate whether the marketing authorization application file is complete and free from any omissions in terms of the requisite data and documents which must be submitted, depending on the type of application and applicable requirements laid down in the Registration Regulation of Human Medicinal Products. The Ministry completes the administrative review and notifies its outcome to the applicant within 30 (thirty) days after receipt of the application file at the Ministry. In the event that deficiencies are identified, the applicant has 30 (thirty) days to address such deficiencies. The second preliminary review, conducted after omissions have been addressed and resubmitted to the Ministry, is completed also within 30 (thirty) days.
In the event that Ministry’s preliminary review finds the applicant to be lacking the requisite qualifications prescribed in the Registration Regulation of Human Medicinal Products,or the file submitted for second preliminary review is again found to be marred by omissions, the application is rejected and returned to the applicant.
According to the Registration Regulation of Human Medicinal Products, the Ministry must complete its regulatory review of the file within 210 (two hundred and ten) days, provided the application file is free from any omissions and has cleared through the preliminary review according to the Registration Regulation of Human Medicinal Products. However, the clock stops for the duration of any extraordinary circumstances or until the applicant submits any data or documents requested by the Ministry, which do not count toward the 210-day timeframe.
The Registration Regulation of Human Medicinal Products lists the following product-related criteria for granting marketing authorization to a human medicinal product: the efficacy of the product has been proven under its intended conditions of use, the safety of the product has been proven, and the product has the appropriate technical and pharmaceutical characteristics. The Ministry may, however, waive some of these criteria taking account of pharmacoeconomic data, when public health considerations warrant it.
Marketing authorization is granted to products which, according to data and documents submitted to and reviewed and analyzed by the Ministry, fulfill the requirements of the Registration Regulation of Human Medicinal Products.
Before offering a product for sale for the first time after receipt of marketing authorization, product samples representative of the final commercial product must be submitted to the Ministry to obtain a “selling permit.” The Ministry reviews the samples for conformity of the package leaflet, package and label information, and price, and grants a selling permit if the product meets the requirements. In view of the above procedural details, the regulatory approval procedure in Turkey is outlined in Figure. 1
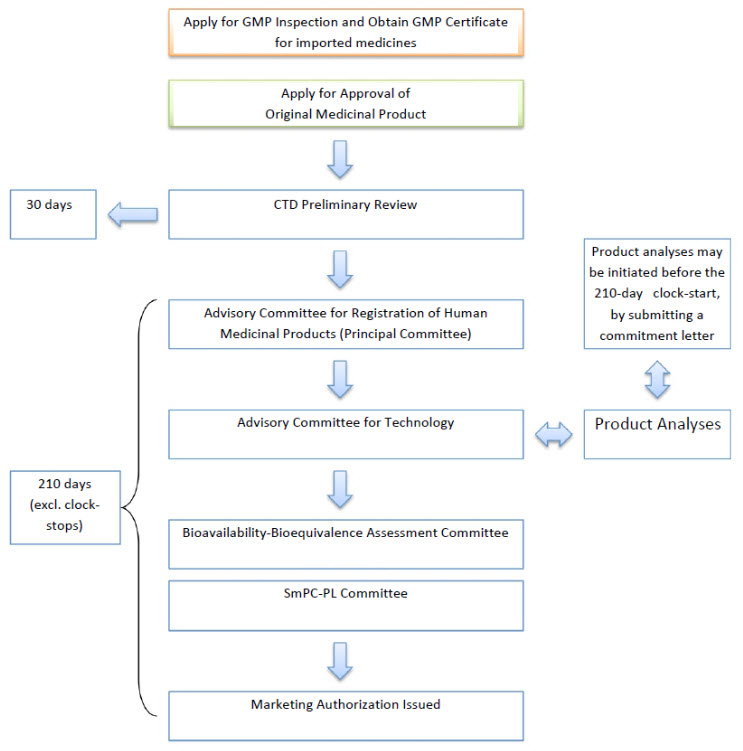
Figure 1: Overview of the Regulatory Approval Procedure in Turkey
New Drug approval process in Canada
Health Canada’s Therapeutic Products Directorate (TPD) regulates pharmaceutical drugs (prescription and nonprescription) and medical devices for human use. Health Canada’s Biologics and Genetic Therapies Directorate (BGTD) is responsible for regulating biologics, including blood and blood products, viral and bacterial vaccines, cells, tissues, organs and xenografts.
Canada’s systems for regulating drug products are very similar to those in the United States. At the federal level, the Therapeutic Products Directorate, an agency of Health Canada that regulates Canada's drug supply, is Canada's counterpart to the FDA. All drug products sold in Canada must be approved by the Therapeutic Products Directorate. Pharmacies in Canada are regulated by the provinces; a similar system to the U.S. in which states regulate pharmacies.
The Canadian pharmaceutical market is the eighth largest in the world, accounting for about two percent of the world market by sales. Canada also has the fourth fastest growing pharmaceutical industry after China, the US and Spain and has shown a steady growth trend.
New drug submission required for new drugs that have not been sold in Canada for a sufficient time and in sufficient quantity to establish their safety and effectiveness—includes clinical trial information and details on production, packaging, labelling, conditions for use, and side effects.
When an NDS is submitted to TPD, it first undergoes an administrative screening procedure to ensure that all necessary parts are included and in the required format. This is not a review of the data. The goal is to complete the screening procedure within 45 days of receipt of the NDS. The file is then directed toward the appropriate Bureau responsible for reviewing drugs in a given therapeutic area. TPD currently has a 300-day performance guideline to complete a standard NDS review, and 180 days to complete a priority NDS.
Terms:
- Notice of Deficiency (NOD): the review cannot continue due to deficiencies or significant omissions in the file.
- Notice of Deficiency: Withdrawal (NOD/w): if the response to an NOD is inadequate, the TPD will issue an NOD/w letter, indicating the company must withdraw the submission.
- Notice of Non-compliance (NON): indicates the review is complete and the submission is deficient or incomplete. It is usually not as severe as an NOD.
- Notice of Non-compliance: Withdrawal (NON/w): if the response to a NON is inadequate, the company must withdraw the submission.
- Notice of Compliance (NOC): once all issues have been resolved, the TPD will issue an NOC. If Health Canada is not satisfied that all issues have been satisfied, the TPD will issue either an NOD/w or NON/w.
- Priority Review: A review status granting eligible new drug submissions and supplements to new drug submissions a shortened review target. This status is granted following review and approval of a request submitted by the sponsor of the drug.
Pre submissions:
- It familiarizes reviewers with the product.
- It identifies the studies on which the sponsor is relying to establish the effectiveness of the drug.
- It provides an opportunity for the sponsor to discuss the submission with Health Canada and obtain feedback.
- Regarding areas of concern or the potential for priority review.
- Meeting requests
- Pre-submission Packages:
Pre-NDS/SNDS meetings:
- A cover letter
- An agenda for the meeting
- A list of specific issues (grouped by discipline) the sponsor would like to discuss or have addressed
- A brief summary of the drug product for which the meeting is being called
- proposed strengths and dosages
- An overview of the market history of the product including the foreign regulatory status of the drug etc.
Submission filing:
- A sponsor files duplicate copies of its submission to the TPD, at which point it undergoes a screening procedure.
- Sponsors seeking a priority review or review under the NOC/c (Notice of Compliance with conditions) regulations should submit a request in advance of filing the NDS.
- For a priority review request, a response from the TPD should be received within 30 calendar days.
- Submission Holds:
- SIPD/CR Holds
- Switch Hold
- Cost-recovery Hold
- Regulatory Hold
Screening:
- The TPD will undertake a screening process to ensure it is complete and in the appropriate format.
- This is an administrative review and does not include any technical review of the information.
- The TPD targets 45 calendar days to complete the screening of an NDSs, SNDSs, ANDSs, SANDSs,
- Once the screening is complete and accepted, the submission enters the queue for technical review.
- If the screening process identifies deficiencies in the NDS, the sponsor will receive a screening deficiency notice, and has 45 calendar days to respond and resolve any identified deficiencies.
Evaluation of Submissions
The TPD has a target of 300 calendar days to complete its evaluation.
- Update Notices
- Requests for Clarification During Screening or Review of the Submission - all submission types
- Notices of Deficiency (NOD) - NDSs, SNDSs, ANDSs, SANDSs, DINAs
- Notices of Noncompliance (NON) - NDSs, SNDSs, SNDS-C, ANDSs, SANDSs, DINAs
Reviewer reports:
The reviewer reports will be provided to the sponsor within seven calendar days following the issuance of an NOD, an NOD/w, an NON or an NON/w.
Sponsors may request a reviewer’s report following the issuance of an NOC, and it is supposed to be provided within 30 calendar days
The regulatory approval procedure in Canada is outlined in Figure. 2
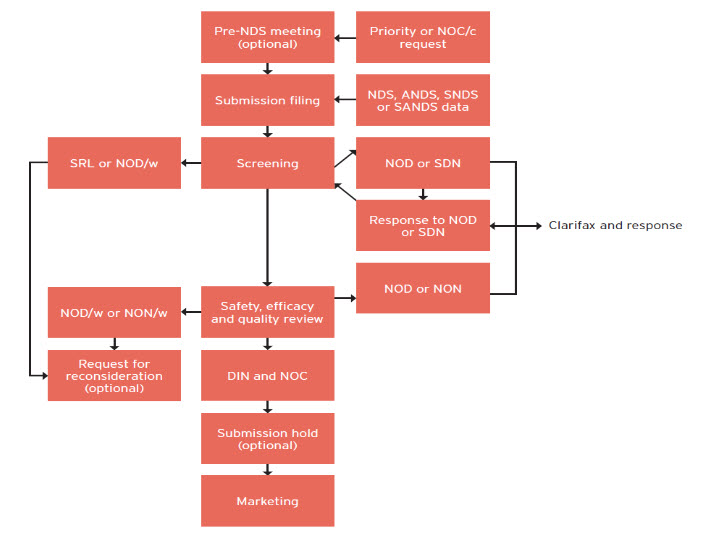
Figure 2: Overview of the Regulatory Approval Procedure in Canada
Regulatory Requirements comparison for Turkey and Canada:
|
S.No |
Filing Information |
Turkey |
Canada |
|
|
1 |
Climatic zone |
Zone 1 & II |
Zone I |
|
|
2 |
Regulatory authority |
Ministry of Health (MOH) |
Therapeutic product directorate (TPD)- For review of ANDS.Health Products and Food Branch Inspectorate (HPFBI)- Plant inspection and Registration of drugs. |
|
|
3 |
Name of the application |
Abridged application. |
Abridged new drug submission (ANDS) |
|
|
4 |
CTD Format |
CTD format implemented |
CTD format implemented |
|
|
5 |
Regulatory partner |
Local agent/ Company in Turkey |
Local agent/ Company in Canada is required |
|
|
6 |
Plant inspection |
GMP certificate by MOH,Turkey / any one of the regulatory authority in European Union (EU) |
GMP certificate by FDA/MHRA/HPFBI |
|
|
7 |
SMF (Site master file) |
Not required for the submission |
Not required for the submission |
|
|
8 |
Patent certificate |
Required for the submission |
Required for the submission |
|
|
9 |
Pharmacopeias |
BP/Ph. Eur. |
BP/Ph. Eur./USP |
|
|
10 |
Language |
English |
English |
|
|
11 |
Front style & Front size |
Times New roman- 12, English |
Times New roman -12, English |
|
|
12 |
Module 2 |
Quality overall summary (QOS) |
Quality overall summary (QOS) |
|
|
13 |
Name of the DMF (Drug master file) |
DMF /CEP LOA is required for DMF from Drug substance supplier |
DMF/CEP LOA is required for DMF from Drug substance supplier |
|
|
14 |
API |
3 different lots of API is required |
2 different lots of API is required. |
|
|
15 |
Submission batches |
3 batches |
2 batches |
|
|
16 |
Batch size |
Two pilot scale and one can be smaller, justified |
2 pilot scale batches |
|
|
17 |
Specifications |
Release and Expiry specification |
Release and Expiry specification |
|
|
18 |
Raw data (Chromatograms, IR spectra, Note books, Calculation sheets) |
Not required for the submission. |
Not required for the submission. |
|
|
19 |
Essential similarity study for Injectables |
Essential similarity study is required to perform with the Innovator sample available from the Turkey/European market. |
Essential similarity study is required to perform with the Innovator sample available from the Canada market. |
|
|
20 |
Batch Product Record and Batch Packaging Record |
|
Required for the submission |
|
|
21 |
Proposed Batch Product Record and Batch Packaging Record |
Required for the submission |
Required for the submission |
|
|
22 |
Process validation |
Process validation protocol and report is required for the submission. |
Process validation protocol and report is required for the submission. |
|
|
23 |
CCS (Container closure system) |
Ph. Eur./BP Pharmacoepialstandard is preferred for the Primary packing material. |
USP/Ph. Eur. Pharmacoepialstandard is preferred for the Primary packing material. |
|
|
24 |
Stability data |
6 Months Accelerated & 6 Months Long term data |
6 Months Accelerated & 6 Months Long term data |
|
|
25 |
Stability conditions |
Long term- 25°C ± 2°C/60% ± 5% Accelerated- 40°C ± 2°C/75% ± 5% |
Long term- 25°C ± 2°C/60% ± 5% Accelerated- 40°C ± 2°C/75% ± 5% |
|
|
26 |
Matrixing/Bracketing |
Encouraged |
Encouraged |
|
|
27 |
Samples |
Samples may/may not require to submit along with the dossier |
Samples are not required to submit along with the dossier |
|
|
28 |
Paper size |
A4 size paper |
8.5 x 11" size paper |
|
|
29 |
BE studies |
Reference product for BE studies should be from the Europe/ Turkey market. |
Reference product for BE studies should be from the Canada market. |
|
|
30 |
Approval time |
8 months |
2 years |
|
Conclusion:
New drugs are an important part of modern medicine. In both countries, information submitted to regulatory authorities regarding the quality, safety and efficacy of drug is similar; however, the time, fee and review process of clinical trials and marketing authorization application differs.For the purpose of harmonization, the International Conference on Harmonisation (ICH) has taken major steps for recommendations in the uniform interpretation and application of technical guidelines and requirements. This step will ultimately reduce the need to duplicate work carried out during the research and development of new drugs. Therefore, harmonization of drug approval processes either by ICH or WHO may be initiated at global level.
Reference:
1. pharmainfo.net/reviews/new-drug-approval-process-regulatory-view
2. Horner A., Comparison of a global submission of new biological entity and a new chemical entity - strategic decisions and criteria for implementation (2005) dra.uni-bonn.de/hoerner. (assessed on March 09th 2010).
3. hastalarin_yenilikci_ilaclara_erisimi_en.pdf
4. Ng R., Drugs: from Discovery to Approval (2004) pharmatext.org/2008/06/drugs-from-discovery-to-approval.html
5. Wang H., Wang C.,Yu W., Hsu S, Huang Y, Lin Y., Leu Y. And Lee C. "From Pharmacovigilance to Pharmacovigilance Planning--The System Building for Safe Medication" Journal of Food and Drug Analysis. 2007; 15:377-386.
6. trade.ec.europa.eu/doclib/html/119478.htm
7. pharmacychecker.com/about-online-pharmacies.asp
8. businesswire.com/news/home/20130416006223/en/Research-Markets-Canada-Pharmaceutical-Industry-2013
9. marsdd.com
10. hc-sc.gc.ca
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








