

{ DOWNLOAD AS PDF }
 ABOUT AUHTORS
ABOUT AUHTORS
Abhijit De*, Chandan Ghosh
Department of Pharmaceutical Science,
Bengal School of Technology, Sugandha,
Hooghly, West Bengal, India
* abhi8981@gmail.com
ABSTRACT
Aging is characterized by a progressive loss of physiological integrity, leading to impaired function and increased vulnerability to death. This deterioration is the primary risk factor for major human pathologies including cancer, diabetes, cardiovascular disorders, and neurodegenerative diseases. In this review, several theories and mechanisms have been put to explain the molecular basis of aging. For example, random damage of the DNA of somatic cell is believed to accumulate with increasing age. Free radicals produced during oxidation of metabolites for energy production also damage DNA and proteins.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2462
|
PharmaTutor (ISSN: 2347 - 7881) Volume 5, Issue 2 Received On: 12/09/2016; Accepted On: 14/12/2016; Published On: 01/02/2017 How to cite this article: De A, Ghosh C; Basics of aging theories and disease related aging-an overview; PharmaTutor; 2017; 5(2); 16-23 |
INTRODUCTION TO AGING
Aging is an extremely complex and multifactorial process that proceeds to the gradual deterioration in functions. It usually manifests after maturity and leads to disability and death. The signs of aging start to appear after maturity, when optimal health, strength and appearance are at the peak. After puberty, all physiological functions gradually start to decline (e.g. the maximum lung, heart and kidney capacities are decreased, the secretion of sexual hormones is lowered, arthritic changes, skin wrinkling, etc). The precise biological and cellular mechanisms responsible for the aging are not known, but according to Fontana and Klein “they are likely to involve a constellation of complex and interrelated factors, including oxidative stress–induced protein and DNA damage in conjunction with insufficient DNA damage repair as well as genetic instability of mitochondrial and nuclear genomes; [1] noninfectious chronic inflammation caused by increased adipokine and cytokine production; [2] alterations in fatty acid metabolism, including excessive free fatty acid release into plasma with subsequent tissue insulin resistanc; [3] accumulation of end products of metabolism, such as advanced glycation end products, amyloid, and proteins that interfere with normal cell function; sympathetic nerve system and angiotensin system activation as well as alterations in neuroendocrine systems; and loss of post-mitotic cells, resulting in a decreased number of neurons and muscle cells as well as deterioration in structure and function of cells in all tissues and organs[4]. In Biological terms, aging represents the molecular biochemical, physiological and structural change that take place in an organism. At all levels of biological organization, there is a progressive decline in adaptation to maintain the homeostatic balance in the functioning of various organs that is characteristic of the adult organism. In general, life span of a multicellular organism is characterized by a smooth transition from the developmental phase to the reproductive phase.
THEORIES OF AGING
There are many theories trying to explain the aging process, each from its own perspective, and none of the theories can explain all details of aging. Some important theories of aging are:
a. Wear and tear theory
This is based on the idea that changes associated with aging result from damage by chance that accumulates over time. The wear-and-tear theories describe aging as an accumulation of damage and garbage that eventually overwhelms our ability to function. The process of aging derives from imperfect clearance of oxidatively damaged, relatively indigestible material and their accumulation hinders cellular catabolic and anabolic functions (e.g. accumulation of lipofuscin in lysosomes)[5].
b. Oxidative stress and free radical theory
Organisms age because of accumulation of free radical induced damage in the cells such as superoxides (O2-), hydrogen peroxide (H2O2), Hydraxyl radicals (OH-), etc. It was subsequently discovered that reactive oxygen species (ROS) generally contribute to the accumulation of oxidative damage to cellular constituents, even though some of them are not free radicals, as they do not have an unpaired electron in their outer shells. Consistently, aged mammals contain high quantities of oxidized lipids and proteins as well as damaged/mutated DNA, particularly in the mitochondrial genome. The oxygen consumption, production of ATP by mitochondria and free-radical production are linked processes [6]. Increases in mitochondrial energy production at the cellular level might have beneficial and/or deleterious effects. Increased regeneration of reducing agents (NADH, NADPH and FADH2) and ATP can improve the recycling of antioxidants and assist the antioxidant defense system. On the other hand, enhanced mitochondrial activity may increase the production of superoxide, thereby aggravating the oxidative stress and further burdening the antioxidant defense system. The mitochondria are the major source of toxic oxidants, which have the potential of reacting with and destroying cell constituents and which accumulate with age. The result of this destructive activity is lowered energy production and a body that more readily displays signs of age (e.g., wrinkled skin, production of lower energy levels). Damaged mitochondria can cause the energy crisis in the cell, leading to senescence and aging of tissue. Accumulation of damage decreases the cell's ability to generate ATP, so that cells, tissues, and individuals function less well. The gradual loss of energy experienced with age is paralleled by a decrease in a number of mitochondria per cell, as well as energy producing efficiency of remaining mitochondria. A major effect of mitochondrial dysfunction is an inappropriately high generation of ROS and proton leakage, resulting in lowering of ATP production in relation to electron input from metabolism. Leaked ROS and protons cause damage to a wide range of macromolecules, including enzymes, nucleic acids and membrane lipids within and beyond mitochondria and thus are consistent with the inflammation theory of aging as being proximal events triggering the production of pro-inflammatory cytokines. Free radicals can damage the mitochondrial inner membrane, creating a positive feedback-loop for increased free-radical creation. Induction of ROS generates mtDNA mutations, in turn leading to a defective respiratory chain. Defective respiratory chain generates even more ROS and generates a vicious cycle.
c. Telomere shortening theory
Telomeres are the strands of DNA that make up the ends of chromosomes. Because of the way in which DNA is replicated, the length of the telomeres shortens each time the cell divides. Consequently, the length of telomeres in the cells of older people tends to be shorter than in younger people. It is thought that, once the telomeres reach a certain minimum size, they can cause the cell to become senescent. In humans, cells can divide approximately fifty times before cell division ceases, presumably as a result of the exhaustion of the telomeres. This is referred to as the ‘Hayflick limit’ after the scientist who first observed it. Telomere shortening has therefore been identified as a factor that could contribute to aging.

Figure 1: Telomeres are DNA caps that sit on the ends of chromosomes. Telomeres shortening and cell replication ceases.
d. The cross linking/Glycation hypothesis of Aging
The cross linking hypothesis is based on the observation that with age, our proteins, DNA, and other structural molecules develop inappropriate attachments or cross-links to one another. These unnecessary links or bonds decrease the mobility or elasticity of proteins and other molecules. Proteins that are damaged or are no longer needed are normally broken down by enzymes called proteases. However, the presence of cross-linkages inhibits the activity of proteases. These damaged and unneeded proteins, therefore, stick around and can cause problems. One of the main ways cross-linking occurs is through a process called glycosylation or glycation. Glucose molecules can stick to proteins, then transform into brownish molecules called advanced glycosylation endproducts, or AGEs. When both of the sticky ends of AGEs adhere to neighboring proteins, they form permanent cross-links that disable the proteins’ functions. Cross-linking of the skin protein collagen, for example, has proven at least partly responsible for wrinkling and other age-related dermal changes Cross-linking of proteins in the lens of the eye is also believed to play a role in age-related cataract formation. Recently, scientists have found evidence that glycation contributes to the formation of beta-amyloid the protein that clumps together in the brains of Alzheimer’s patients. Carnosine occurs in very low concentrations in the brain and other tissues. In the laboratory carnosine has been shown to delay the senescence or aging of human cells called fibroblasts. Carnosine works by preventing cross-linking of proteins.
e. The genome maintenance hypothesis/Somatic mutation
Damage to our DNA happens thousands of times every day in every cell in our body throughout our lives. This damage can be caused by oxidative free radicals, mistakes in replication, or outside environmental factors such as radiation or toxins. Mutations or spontaneous changes in the structure of our genes that occur in our egg or sperm cells will be passed on to future generations, if those mutations are not so potentially disruptive as to be fatal to our offspring. Mutations that occur in the rest of the cells of the body will only affect that individual and cannot be passed on to future generations. Most of those body cell, or somatic, mutations will be corrected and eliminated, but some will not. Those will accumulate, eventually causing the cells to malfunction and die. This process, it has been suggested, is a crucial component in the aging process. This theory also encompasses a role for mitochondria, the cellular powerhouses, as important factors in aging. Mitochondria create damaging free radicals as a by-product of normal energy production. Somatic mutations in the DNA of the mitochondria accumulate with age, increasing free radical production, and are associated with an age-related decline in the functioning of mitochondria.
f. Neuroendocrine hypothesis of aging
The Neuroendocrine system refers to the complex connections between the brain and nervous system and our endocrine glands, which produce hormones. The hypothalamus, a structure at the base of the brain, stimulates and inhibits the pituitary gland, often called the “master gland,” which in turn regulates the glands of the body (ovaries, testes, adrenal glands, thyroid) and how and when they release their hormones into our circulation. As we age, this system becomes less functional, and this can lead to high blood pressure, impaired sugar metabolism, and sleep abnormalities. The effects that the various hormones our different glands produce have on different facets of aging have been studied extensively.
g. The evolutionary senescence theory of aging-
The most widely accepted overall theory of aging is the evolutionary senescence theory of aging. Unlike the earlier programmed theory of evolution and aging, which tried to find reasons why evolution might favor aging, evolutionary senescence theory focuses on the failure of natural selection to affect late-life traits. Natural selection, because it operates via reproduction, can have little effect on later life. Genes and mutations that have harmful effects but appear only after reproduction is over do not affect reproductive success and therefore can be passed on to future generations. In 1952, Peter Medawar proposed that the inability of natural selection to influence late-life traits could mean that genes with detrimental late-life effects could continue to be passed from generation to generation. This theory is called the mutation accumulation theory. A few years later, George Williams extrapolated on this idea by formulating the theory of “antagonistic pleiotropy.” Antagonistic pleiotropy means that some genes that increase the odds of successful reproduction early in life may have deleterious effects later in life. Because the gene’s harmful effects do not appear until after reproduction is over, they cannot be eliminated through natural selection. An example of antagonistic pleiotropy in humans is p53, a gene that directs damaged cells to stop reproducing or die. The gene helps prevent cancer in younger people, but may be partly responsible for aging by impairing the body’s ability to renew deteriorating tissues. Because of antagonistic pleiotropy, it is likely that tinkering with genes to improve late-life fitness could have a detrimental effect on health at younger ages.
h. Mitohormesis theory of aging
This theory is based on the “hormesis effects”. It describes beneficial actions resulting from the response of an organism to a low-intensity stressor. It has been known since the 1930s that restricting calories while maintaining adequate amounts of other nutrients can extend the lifespan in laboratory animals. Michael Ristow's group has provided evidence for the theory that this effect is due to increased formation of free radicals within the mitochondria causing a secondary induction of increased antioxidant defense capacity. The best strategy to enhance endogenous antioxidant levels may actually be oxidative stress itself, based on the classical physiological concept of hormesis[7].
i. Mitochondrial theory of aging
Mitochondria are the cell’s chief energy producing organelles. A cell can contain hundreds of mitochondria, the DNA of which encodes a subset of mitochondrial RNA and proteins. The mitochondrial theory of ageing proposes that mutations progressively accumulate within the mitochondrial DNA, leading to a cellular ‘power failure’[8]. The consequences are predicted to be particularly dire for non-proliferative cells in organs that have a minimal capacity to regenerate (quiescent tissues), such as the heart and brain. The activity of master regulators of mitochondrial function and number diminishes with ageing, further contributing to mitochondrial deficiency. With age, telomere damage in the nucleus triggers the activation of p53, which can have different effects. In proliferative cells, p53 halts both cell growth and DNA replication, potentially causing apoptotic cell death. p53 also represses the expression of PGC-1 in mitochondria, reducing the function and number of these organelles, and so leading to age-related dysfunction of mitochondrion-rich, quiescent tissues. The mitochondrial derangements driven by loss of PGC-1 activity may independently lower the threshold for the generation of toxic intermediates such as reactive oxygen species (ROS), which damage mitochondrial DNA, thus setting up a vicious cycle of further mitochondrial dysfunction[9].
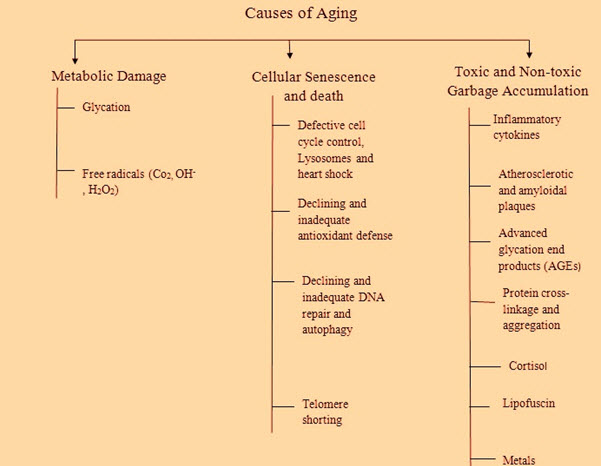
Figure 2: Common causes of aging
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
DISEASES INDUCED AGING
Aging is the major risk factor for the predominant killer disease of developed countries, including dementia, cardiovascular disease and cancer etc.
a. Telomere and age related disease
It is well established that telomere length and telomerase activity are important factors in the pathogenesis of human diseases[10]. Telomerase activation may prove to be useful in the treatment of chronic and degenerative diseases associated with telomere loss We summarize information on the age-related diseases below, which have been proposed to be related to telomeres.
i. Cardiovascular diseases
Atherosclerosis is also an aging-related systemic disease. Telomere length is a new marker of cardiovascular risk. In the vascular endothelium, shorter telomeres are found in those areas of the arterial wall that are more susceptible to atherosclerosis because of higher haemodynamic stress. It is believed that this stress results in a more rapid cell turnover In addition, vascular endothelial cells and smooth muscle cells in the vicinity of atherosclerotic lesions often stain positive for the activity of β-galactosidase, which is a rather nonspecific marker for cellular senescence[11]. Furthermore, the induction of telomere dysfunction in endothelial cells in vitro generates a senescent phenotype with an atherogenic protein expression profile. Healthy individuals with shorter telomere were likely to develop hypertension, and hypertensive individuals with shorter telomere were more susceptible to develop atherosclerosis. Heart failure is a frequent cause of death in the aging human population. Telomere shortening with age might also contribute to cardiac failure in humans, opening the possibility for new therapies. Chronic heart failure is characterized by increased myocyte apoptosis and telomere erosion. In humans, the formation of myocytes from telomerase-positive cardiac stem cells appears to be necessary for cardiac homeostasis.
ii. Diabetes
Type 2 diabetes is caused by a combination of peripheral insulin resistance and b-cell dysfunction. Telomeres were significantly associated with type 2 diabetes, which could be partially attributed to the high oxidative stress in the patients with type 2 diabetes. Moreover, short telomeres are predictors of progression of diabetic nephropathy and of all-cause mortality in the patients with type 1 diabetes.
iii. Cancer
The classical telomere hypothesis suggests that the telomere shortening provides a tumor suppressor mechanism to cease the growth of transformed cells. In normal somatic cells, the absence of telomerase can lead to telomere shortening and cell senescence. However, the majority of cancer cells exhibit a high telomerase activity. The reappearance of telomerase activity is triggered by as yet unclear molecular mechanisms and enables cancer cells to maintain telomere length. Studies have revealed a dual role of telomere shortening in carcinogenesis. On the one hand, shortened telomeres induce chromosomal instability, which is the most important cause of cancer initiation during aging. On the other hand, telomere stabilization is required for tumor progression. The initiation of chromosomal aberrations by telomere shortening might contribute to the increased cancer rate of cancer onset during aging. Additionally, telomere shortening may initiate the tumor genesis of gastric carcinoma. The ongoing basic research in this field helps to constantly define new targets and to increase the specificity, safety and efficacy of existing therapeutic approaches directed against telomeres and telomerase. Therefore, therapies targeting telomerase with telomerase inhibitors revealed the potential for cancer therapy. Recent research developments for each of the anti-telomerase approaches including antisense-oligonucleotides, hammerhead ribozymes, dominant negative hTERT, reverse-transcriptase inhibitors, immunotherapy, G-quadruplex stabilisers, gene therapy, small molecule inhibitors and RNA interference have been provided[12].
iv. Immune system diseases
The immune system is a prime example of a highly dynamic cellular system, for which telomere maintenance is pivotal. Immune competence is strictly dependent on rapid expansions of clonal T- and B-cell populations, and telomere loss may contribute to defective immune responses in the elderly. Equally interestingly, accelerated T-cell aging combined with telomeric shortening may prompt an autoimmune response and thereby explain the increased susceptibility for chronic inflammatory diseases in the elderly. Recent reports have suggested that telomere shortening is involved in the dramatic age-related alterations of the immune system, and this is considered one of the major factors affecting morbidity and mortality. The gradual decline with age in the capacity of the immune system to recognize and eliminate antigens is an important causal factor for many diseases in the elderly. Although both the innate and adaptive immune responses are weakened in old people, it is the decline in T-cell action that proves to have the most injurious effect on the elderly. T cells become clonally exhausted which leads to a decline in the adaptability of the response of the immune system[13]. The senescence of T cells in vivo is well documented: as the number of population doublings in culture increases then there is a progressive decrease in the proportion of T cells that express CD28. This results in an inability of the T cells to proliferate and a reduction in telomerase induction by CD28, as well as a resistance to apoptosis. In addition, research on bone marrow transplantation has provided important clues for the in vivo interactions of immunosenescence and telomere length and consequences.
v. Dyskeratosis congenital
Defective telomere function or mutations in the DNA repair system can induce some human disorders associated with shorter telomere length, such as dyskeratosis congenital (DC)[14]. Most direct evidence for telomere shortening during human aging comes from research on DC, which is a premature aging disease characterized by telomerase and telomere dysfunction. Autosomal dominant dyskeratosis congenital is associated with mutations in the RNA component of telomerase, while X-linked dyskeratosis congenital is due to mutations in the gene encoding dyskerin.
b. Alzheimer’s disease
Brain damage and the death of brain cells affect brain function and cognitive ability. The prevailing paradigm of Alzheimer’s disease pathogenesis contends that the primary pathogenic event is the extraneuronal or intraneuronal, or both, accumulation of the misfolded protein, amyloid beta-peptide which initiates a pathogenic cascade resulting in neurotoxicity and ultimately the clinical syndrome of Alzheimer’s disease. This paradigm has its origins in the autosomal dominant forms of the disease, which account for 1–2% of all cases. These inherited forms are associated with mis-sense mutations of genes that encode the amyloid precursor protein (APP), or proteolytic enzymes that cleave APP (eg, presenilin 1 and 2). Such mutations are associated with an increased production of amyloid beta peptide and result in early-onset of aging.
c. Parkinson’s Disease
Parkinson’s disease is a progressive neurodegenerative disease, the second most common disorder of this type after Alzheimer’s disease. It progresses slowly as small clusters of dopaminergic neurons in the midbrain die. The gradual loss of these neurons results in reduction of a critical neurotransmitter called dopamine, a chemical responsible for transmitting messages to parts of the brain that coordinate muscle movement. Common motor symptoms are tremors or shaking in hands, arms, legs, jaw, and face; rigidity or stiffness of limbs and trunk; bradykinesia, or slowness of movement; and difficulties with balance, speech, and coordination. Symptoms begin gradually and typically worsen over time. There is also a collection of nonmotor symptoms, such as poor sense of smell, constipation, depression, cognitive impairment, fatigue, and other impairments that also accompany Parkinson’s. Some of these symptoms may develop years before the onset of motor problems.

Figure 3: Parkinson’s patients have less dopamine
d. Premature Aging
Premature aging, called also accelerated aging, is a group of genetic syndromes, in which the children have premature aging. The three known premature aging syndromes of human being are Hutchinson–Gilford Progeria Syndrome (HGPS), Werner syndrome (WS), and Cockayne syndrome (CS). These syndromes have differences on genetic backgrounds, on ages of onset of abnormity, and on symptoms; however they are common on typical aging changes, including hair loss, tooth loss, thinness and hardness of skin, skin wrinkles, and senior spots. The abnormality in tissue structure is the common point between premature aging and normal aging, and it links a defective development and a defective repair, the Misrepair. Defective development is a result of mis-construction of tissues/organs, as a consequence of genetic mutation; whereas aging is a result of mis-reconstructions, the Misrepairs, for maintaining the structure of tissues/organs. Construction-reconstruction of the structure of an organism is thus the coupling point between development and aging. Mis-construction and Mis-reconstruction (Misrepair) are the essential processes in the development of aging-like feathers. Misrepair is defined as incorrect reconstruction of an injured living structure such as a molecule, a cell and a tissue.
Hutchinson–Gilford Progeria Syndrome (HGPS) is the first syndrome that is named as premature aging syndrome or Progeria. HGPS is a genetic condition with abnormal development and appearance of premature aging features from infancy[15]. The Progeria children often look normal at birth; however they manifest the abnormal growth after birth. Growth abnormalities develop progressively with age, including growth failure, hair loss, hardening and thinness of skin, wrinkled skin, stiff joints, atherosclerosis, and loss of body fat and muscle. HGPS children all have a small body but big head with prominent eyes and narrow face, and they die mainly from heart attack or stroke in young ages. A gene mutation on lamin A (LMNA gene), a protein for composing nuclear lamin, is identified in the HGPS patients. Werner syndrome (WS), called also “adult progeria”, is a genetic disorder characterized by an early and progressive development of aging features[15]. A mutation on WRN gene was identified in the WS patients. Normal WRN protein is a kind of DNA helicase, and it assists DNA duplication and maintains the functionality of telomeres.
Cockayne syndrome (CS), called also “Weber-Cockayne syndrome” and “Neill-Dingwall syndrome”, is a rare genetic condition characterized by growth failure, impaired development of neural system, abnormal sensitivity to sunlight, and appearance of premature aging.
CONCLUSION
Aging is a syndrome of changes that are deleterious, progressive universal and thus for irreversible. Several theories have been applied in support of aging. It is a cellular process, or a genetic process that every individual has to be gene through. It cannot be stopped at all but can be reduced the rate by various therapies. Recently various approaches are emerged towards the preventer of aging. In recent years various advancements in this field has be noted. Branded products have a good success rate that referral product. Though now days, polyherbal technology has offered a new light in anti-aging treatment. Moreover disease induced aging are more prevalent with lot of disorders. So disease specific drugs are formulated to have few roles as anti-aging therapy. Novel approaches such as Rapalogs and sirtuin1 analogs have brought a promising contribution as noted for preclinical data. Clinical studies are now being conducted at several centers to figure at potential role of new molecules with less toxic effects. So, it can be concluded that our review clearly explain the theory and etiology behind aging and their possible treatments either clinically or by some others means.
REFERENCES
1. Martin G.M., Austad S.N. and Johnson T.E; Genetic analysis of ageing: Role of oxidative damage and environmental stress; Nat. Genet; 1996; 13; 25-34.
2. Davies K.J; Oxidative stress: the paradox of aerobic life; Biochem. Soc. Symp; 1995; 61; 1-31.
3. Martin G.M; Interaction of aging and environmental agents: The gerontological perspective; Prog. Clin. Bio. Res; 1987; 228; 25-80.
4. Gilca M., Stoian I., Atanasiu V. and Virgolici B; The oxidative hypothesis of senescence; J. Postgrad. Med; 2007; 53; 207-213.
5. Harman D; Aging: a theory based on free radical and radiation chemistry; J. Gerontol; 1956; 11; 298-300.
6. Sohal R; Role of oxidative stress and protein oxidation in the aging process; Free. Radic. Biol. Med; 2002; 33; 37-44.
7. Schulz T.J., Zarse K., Voigt A., Urban N., Birringer M. and Ristow M; Glucose Restriction Extends Caenorhabditis elegans Lifespan by Inducing Mitochondrial Respiration and Increasing Oxidative Stress; Cell. Metab; 2007; 6; 280-293.
8. Hagen T.M; Oxidative stress, redox imbalance, and the aging process; Antioxid. Redox. Signal; 2003; 5; 503-506.
9. Yang J.H., Lee H.C., Lin K.J. and Wei Y.H; A specific 4977- bp deletion of mitochondrial DNA in human aging skin; Arch. Dermatol. Res; 1994; 286; 386-390
10. Shammas M.A; Telomeres, lifestyle, cancer, and aging; Cur. Opinion. Clin. Nutr. Metab. Care; 2011; 14; 28–34.
11. Yang Z.W., Huang X., Jiang H., Zhang Y.R., Liu H.X., Qin C. and Eisner G.M; Short telomeres and prognosis of hypertension in a chinese population; Hypertension; 2009; 53; 639–695.
12. Agrawal A., Dang S. and Gabrani R; Recent patents on anti telomerase cancer therapy; Recent. Pat. Anticancer. Drug. Discov; 2012; 7; 102–117.
13. Akbar A.N., Beverley P.L. and Salmon M; Opinion: will telomere erosion lead to a loss of T-cell memory?; Nat. Rev. Immunol; 2004; 4; 737–743.
14. Mitchell J.R., Wood E. and Collins K; A telomerase component is defective in the human disease dyskeratosis congenital; Nature; 1999; 402; 551–555.
15. Katayama K., Armendariz-Borunda J., Raghow R., Kang A.H. and Seyer J.M; A pentapeptude from type I collagen promotes extracellular matrice production; J Biol.Chem; 1993; 268; 9941-9944.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






