

 About Author:
About Author:
*1 Patel Dimpalben Girishkumar, 2 Mr.K.H.Shah, 3 Rohit K Patel, 3Yatish Shukla, 3 Modi B, 3 Nilesh Patel
1 Dharmaj Degree Pharmacy College,
Dist- Anand, Dharmaj -388430, Gujarat
2 Professor, IPCPRC, Dharmaj, Gujarat
3 KAPTAB Pharmaceuticals
*dimplepatel70@gmail.com
Abstract
The aim of the current investigation is to design oral once daily modified release dosage forms of amoxicillin trihydrate for treatment of pharyngitis/tonsilitis, which release the drug for 24 hours and match with theoretical drug release profile. The tablets and capsules were prepared by the different method using different polymers in different concentrations. The interference of the polymers was ruled out by FT-IR spectroscopy studies. The powder-blends of tablets and drug were evaluated for their physical properties like angle of repose, bulk density, compressibility index, and Hausner ratio and found to be satisfactory. The manufactured tablets were evaluated for in process and finished product quality control tests including appearance, thickness, weight variation, hardness, friability, drug content, and in vitro drug release. All formulations showed appearance, thickness, weight variation, hardness, friability and drug content in specified limit. All formulations showed acceptable pharmacotechnical properties and complied with in-house specifications for tested parameters. The results of dissolution studies indicated that formulation containing 50% ethyl cellulose and 50% methocel was the most successful formulation which was evidenced by similarity (f2) and dissimilarity (f1) factors. The formulated amoxicillin trihydrate tablets followed zero order release kinetics and Higuchi diffusion was the dominant mechanism of drug release, resulting in regulated and complete release within 24 hours. Formulations were subjected to short term stability studies as per ICH guidelines and were found stable. Capsule formulations 16 were evaluated for weight uniformity, drug content and in vitro drug release. The results of dissolution studies indicated that drug release from capsule not extend up to 24hrs. All formulations of capsule failed in in-vitro drug release test. In comparison of tablet and capsule formulations, tablet found to be successful dosage form.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1483
Introduction
Conventional dosage forms, which are prompt release in nature, have been used from decades as antibiotics for different infection. To maintain drug concentration within the therapeutically effective range, it is necessary to take these types of dosage forms several times a day and which results in the fluctuations in drug levels. Recently, several technical advancements have been made which results in new techniques for drug delivery. These techniques are capable of controlling the rate of drug delivery, extending the duration of therapeutic activity and / or targeting the delivery of drug to a tissue. Modified release pharmaceutical dosage forms may offer one or more advantages over conventional dosage forms of the same drug. Modified release dosage forms continue to draw attention in the search for improved patient compliance and decreased incidences of adverse drug reactions and decrease total dose of drug. Ideally, a extended release dosage form will provide a therapeutic concentration of the drug in te blood that is maintained throughout the dosing interval with a reduction in a peak concentration ratio. One of the least complicated approaches is to form a tablet and capsule. Various types of polymers used for modified release of drug from dosage form and their modeling aspects have been reviewed. It contained general concepts and requirements for modified release drug delivery system. Classification, advantages and disadvantages of oral extended release drug delivery systems, ideal characteristics of extended release formulations, concept of tablet and capsule, and overview about antibiotic and disease were discussed.
Objectives
The objectives of the research work undertaken are;
1) To prepare different modified release dosage form of amoxicillin for the treatment of pharyngitis/tonsillitis.
2) To study the Preformulation factors such as melting point, drug-excipients interaction, angle of repose, carr’s index, drug property etc.
3) To characterize manufactured tablets for hardness, thickness, content uniformity, weight uniformity, dimensions, etc. and capsule for weight uniformity, content uniformity, appearance.
4) To study in vitro drug release study comparison of different dosage form like tablet, capsule.
5) To study in vitro drug release study of different dosage form with marketed product in US or with the ideal theoretical drug release profile.
6) To carry out short term accelerated stability studies on the most satisfactory formulation as per ICH guidelines.
In the present investigation, efforts were made to develop modified release tablets and capsule of amoxicillin trihydrate for treatment of Pharyngitis/Tonsilitis, which will provide similar in vitro release profiles to that of developed theoretical drug release profiles which can be confirmed by calculating f1 (difference factor) and f2 (similarity factor) values.
[adsense:468x15:2204050025]
1.1 MODIFIED RELEASE DOSAGE FORM
Modified preparations where the rate and/or place of release of the active ingredient are different from that of the conventional dosage form administered by the same route. This deliberate modification is achieved by special formulation design and/or manufacturing method. Modified release dosage forms include prolonged release, delayed release, pulsatile release and accelerated release dosage forms.1
Modified release dosage form is the dosage forms whose drug-release characteristics of time course and/or location are chosen to accomplish therapeutic or convenience objectives not offered by conventional dosage forms such as a solution or an immediate release dosage form. Modified release solid oral dosage forms include both delayed and extended release drug products.2
Drug products designed to reduce the frequency of dosing by modifying the rate of drug absorption have been available for many years. Early modified-release products were often intramuscular/subcutaneous injections of suspensions of insoluble drug complexes, e.g. procaine penicillin, protamine zinc insulin. Advances in technology have resulted in novel oral modified-release dosage forms. Many terms are used to describe modified-release products including extended-release, prolonged-release, controlled-release, controlled-delivery, slow-release and sustained-release. These preparations, by definition, have a reduced rate of release of active substance.3
Oral solid dosage forms are the preferred route for many drugs and are still the most widely used formulations for new and existing modified release (MR) products.
Over many years, approaches and technologies in the area of MR oral drug delivery have been developed to:
* Extend the release of drug over a number of hours, an effect accomplished either by combining the drug with release-retardant materials to form a matrix core, or applying release-modifying film coatings to cores containing the drug.
* Delay the release of drug for a period of time, usually through the application of an externally applied enteric coating.
Technologies are available for the formulation, development and production of MR tablets and multiparticulates such as drug-loaded pellets and granules, mini-tablets and drug crystals.
Over the last decade, the approach to MR oral drug delivery systems has changed from a line extension strategy to a clinically superior approach for marketed drugs as well as for new chemical entities. The benefits offered by MR systems include reduced dosing frequency with improved patient compliance, better and more uniform clinical effects with lower incidence of side effects and possible enhanced bioavailability. The rational design of MR systems, where biological, physicochemical and physicomechanical considerations have been taken into account during formulation of MR dosage form, has alleviated the risk of ‘dose dumping’ in vivo. In addition to the pharmacological and patient benefits, MR dosage forms offer commercial opportunity through intellectual property, brand differentiation and recognition, plus the potential to license technologies to other companies.
The United States Pharmacopoeia definition of an MR system is that: “the drug release characteristics of time, course and/orlocation are chosen to accomplish therapeutic orconvenience objectives not offered by conventionaldosage forms...”
This includes technologies that modify the site of drug delivery. The successful formulation of an MR device requires a comprehensive understanding of the mechanisms of drug release from the macroscopic effects of size, shape and structure through to chemistry and molecular interactions. Multiparticulate dosage forms have been shown to be less prone to food effects than monolithics1 and are often the preferred formulation for extended and/or delayed release. Film coating is an ideal process for the production of extended release multiparticulate dosage forms. For application in extended release delivery systems, film coats with well-characterised permeability properties are essential.
An important MR technology is delayed release through application of gastro-resistant coatings. In this case, a coating layer is applied to the dosage form, either multiparticulate or monolithic, providing protection to the stomach from the drug or protecting the drug from exposure to acidic gastric fluids. The majority of modern enteric coatings rely on polymers containing carboxylic acid groups as the functional moiety. These groups remain unionised in the low pH environment of the stomach but start to ionise as the dosage form passes into the small intestine. As the pH level rises above the point of dissolution, the polymer is ionised and the drug is released. In the past, enteric coating systems have required the use of non-aqueous solvents for application; however, the majority of new enteric coating developments are based on aqueous enteric polymeric systems.4
1.1.1 IMMEDIATE RELEASE
The term “immediate release” pharmaceutical formulation includes any formulation in which the rate of release of drug from the formulation and/or the absorption of drug, is neither appreciably, nor intentionally, retarded by galenic manipulations. In the present case, immediate release may be provided for by way of an appropriate pharmaceutically acceptable diluent or carrier, which diluent or carrier does not prolong, to an appreciable extent, the rate of drug release and/or absorption.5
1.1.2 EXTENDED RELEASE
Extended release is designed to slow the rate of release of the active ingredient(s) in the gastrointestinal tract.Some extended-release medications have the letters "XL" or "LA" or "XR" in their name.Extended-release medications have special coatings or ingredients that control how fast the drug is released from the dosage form into body. This may allow you to take certain medications only once a day, instead of more often.
The extended release formulations are the type of formulations which will improves the therapeutic index of drug concentration. This formulation makes the drug available over extended time period after oral administration.6
Advantages6
* Sustained Blood Levels:
The size and frequency of dosing is determined by the pharmacodynamics and pharmacokinetic properties of the drug. The slower the rate of absorption, the less the blood concentrations fluctuate within a dosing interval. This enables higher doses to be given less frequently. For drugs with relatively short half-lives, the use of extended release products may maintain therapeutic concentrations over prolonged periods.
* Attenuation of adverse effects:
With conventional dosage forms, high peak blood concentrations may be reached soon after administration. With possible adverse effects related to the transiently high concentration. An example is hypotension in patients taking rapid-release nifedipine products. The use of an extended-release product avoids the high initial blood concentrations which cause the sudden reduction in blood pressure and other significant hemodynamic changes such as reflex tachycardia. Anotherexamples are the transient nausea at sub-toxic concentration of some conventional release products such as throphylline.
* Improved Patient Compliance:
Drugs with short half-lives often need to be given at frequent intervals to maintain blood concentrations within the therapeutic range. There is an inverse correlation between the frequency of dosing and patient compliance. A reduction in the number of daily doses offered by extended-release products has the potential to improve compliance. However, this advantage probably only occurs when conventional formulations need to be given three or more times a day.
* The extended release formulations may maintain therapeutic concentrations over prolonged periods.
* The use of extended release formulations avoids the high blood concentration.
* Extended release formulations have the potential to improve the patient compliance.
* Reduce the toxicity by slowing drug absorption.
* Increase the stability by protecting the drug from hydrolysis or other degradative changes in gastrointestinal tract.
* Minimize the local and systemic side effects.
* Improvement in treatment efficacy.
* Minimize drug accumulation with chronic dosing.
* Usage of less total drug.
* Improvement the bioavailability of some drugs.
* Improvement of the ability to provide special effects. E.g.: Morning relief of arthritis through bed time dosing.
Disadvantages6
* High cost of preparation.
* The release rates are affected by various factors such as, food and the rate transit through the gut.
* Some differences in the release rate from one dose to another dose but these have been minimized by modern formulations.
* Extended release formulation contains a higher drug load and thus any loss of integrity of the release characteristics of the dosage form.
* The larger size of extended release products may cause difficulties in ingestion or transit through gut.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
1.1.2.1 Ideal Characteristics of the Drug Candidate for Extended Release Formulation6
A. Physiochemical Properties of the drug:
a) Aqueous solubility:
Lower limit solubility for such product is reported to be 0.1 mg/ml. As the drug must be in solution form before absorption, drug having low aqueous solubility usually suffers oral bioavailability problem due to limited GI transit time of undissolved drug and limited solubility at absorption site. So these types of drug are undesirable.
Drug having extreme aqueous solubility are undesirable for ER because, it is too difficult to control release of drug from the dosage form.
Physiological pH dependent solubility i.e. variation in solubility at different GI pH are undesirable (e.g. Aspirin, which is less soluble in stomach, but more soluble in intestine) as it will yield variation in dissolution rate. A drug with good aqueous solubility, pH independent solubility is desirable for oral new drug delivery system
b) Partition Co-efficient:
As biological membrane is lipophilic in nature through which the drug has to pass though, so partition co-efficient of drug influence the bioavailability of drug very much. Drug having lower partition co-efficient values less than the optimum activity are undesirable for oral ER drug delivery system, as it will have very less lipid solubility and the drug will be localized at the first aqueous phase it come in contact e.g. Barbituric acid.
Drug having higher partition co-efficient value greater than the optimum activity are undesirable for oral ER drug delivery system because more lipid soluble drug will not partition out of the lipid membrane once it gets in the membrane. The value of partition co-efficient at which optimum activity is observed is approximately 1000:1 in 1-octano/water system.
c) Drug stability in-vivo:
As most of ER Drug delivery system is designing to release drug over the length of the GIT, hence drug should be stable in GI environment. So drug, which is unstable, can’t be formulated as oral ER drug delivery system, because of bioavailability problem.
E.g. - Nitroglycerine.
d) Protein binding:
The Pharmacological response of drug depends on unbound drug concentration drug rather than total concentration and all drug bound to some extent to plasma and or tissue proteins. Proteins binding of drug play a significant role in its therapeutic effect regardless the type of dosage form as extensive binding to plasma increase biological half life and thus sometimes ER drug delivery system is not required for this type of drug.
e) Drug pKa& Ionization at physiological pH:
As we know only unionized drug are absorbed and permeation of ionized drug is negligible, since its rate of absorption is 3 to 4 times less than that of the unionized drug. pKa range for acidic drug where ionization is pH sensitive is around 3.0 – 7.5 and pKa range for basic drug whose ionization is pH sensitive is around 7.0-11.0 are ideal for optimum positive absorption. Drug shall be non-ionized at the site to an extent 0.1 – 5.0%. Drugs existing largely in ionized form are poor candidates for oral ER drug delivery system. e.g.:- Hexamethonium.
f) Mechanisms and sites of absorption:
Drug absorption by carrier mediated transport and those absorbed through a window are poor candidate for oral ER drug delivery system e.g. – several B vitamins. Drugs absorbed by passive diffusion, pore transport and through over the entire length of GIT are suitable candidates for oral ER drug delivery system.
g) Molecular size and diffusivity:
With large molecular size are poor candidate for oral ER drug delivery system because it the ability of the drug to diffuse polymeric membrane is a function of its diffusivity (or diffusion co-efficient). Diffusivity depends on size shape of the cavities of the membrane. The diffusion co-efficient of intermediate molecular weight drug i.e.-100 to 400 Dalton, through flexible polymer range from 10-6 to 10-9 cm2/sec. For drugs having molecular weight > 500 Daltons the diffusion co-efficient in many polymers are very less i.e. less than 10-12 cm2/sec. Drugs is very difficult to control release rate of medicament from dosage form e.g. proteins and peptides.
h) Dose size:
If a product has dose size >0.5gm it is a poor candidate for oral ER drug delivery system, because increase in bulk of the drug, thus increases the volume of the product.
B. Biological Properties of Drug: -
a) Absorption:
For oral ER drug delivery system the rate of drug absorption (ka) should be more -API than that of the rate of drug release (kr) from the dosage form i.e. kr<<<ka. Drug that are slowly absorbed or absorbed with a variable absorption rate of elimination of drug are poor candidate for oral ER drug delivery system. Some possible reasons for a low extent of absorption are poor water solubility, small partition co-efficient, acid hydrolysis, and metabolism or its site of absorption.
b) Distribution:
Drugs with high apparent volume of distribution, which influence the rate of elimination of the drug, are poor candidate for oral ER drug delivery system e.g. Chloroquine.
c) Metabolism:
Drug, which extensively metabolized is not suitable for ER drug delivery system. A drug capable of inducing metabolism, inhibiting metabolism, metabolized at the site of absorption of first-pass effect is poor candidate for ER delivery, since it could be difficult to maintain constant blood level e.g. levodopa, nitroglycerine.
d) Half-life of drug:
A drug having biological half-life between 2 to 8 hours is best suited for oral ER drug delivery system. As if biological half-life < 2hrs the system will require unacceptably large rate and large dose and biological half-life >8hours formulation of such drug into oral ER drug delivery system is unnecessary.
e) Margin of safety:
As we know larger the value of therapeutic index safer is the drug. Drugs with less therapeutic index usually poor candidate for formulation of oral ER drug delivery system due to technological limitation of control over release rates.
f) Plasma concentration response relationship:
Generally pharmacological response of drug depends on plasma drug concentration rather than size and dose. But some drugs pharmacological activity is independent of plasma concentrations, which are poor candidate for oral ER drug delivery system. E.g. Reserpine.
g) Concentration dependency on transfer of drug:
Transfer of drug from one compartment to other by zero kinetic process then such drugs are poor candidate for oral ER delivery system, it should be first order kinetics.
1.1.2.2 Types of Extended Release Formulation6
Many current oral extended release systems are available
a) Dissolution-controlled release system.
b) Diffusion-controlled release system.
c) Osmotic pump system.
d) Erosion controlled release systems.
a) Dissolution controlled release systems:
In dissolution controlled extended release systems the rate of dissolution in the gastrointestinal juices of the drug or another ingredients is the release controlling process. Sparingly water-soluble drug can form a preparation of a dissolution controlled extended release type. Reduced drug solubility can be accomplished by preparing poorly soluble salts or derivatives of the drug. An alternative means to achieve extended release based on dissolution is to incorporate the drug in a slowly dissolving carrier.
Dissolution controlled extended release systems can also be obtained by covering drug particles with a slowly dissolving coating. The release of the drug from such units occurs in two steps,
1. The liquid that surrounds the release unit dissolves the coating (rate limiting dissolution step).
2. The solid drug is exposed to the liquid and subsequently dissolves sustained release oral products employing dissolution as the rate limiting step are in principle the simplest to prepare.
A drug with a slow dissolution rate is inherently sustained. Some example of these drugs includes digoxin, griseofulvin, and salicylamide. Others, such as aluminum aspirin, ferrous sulfate, and benzphetaminepaomate, produce such forms when in contact with the absorption pool contents.
For those drugs with high water solubility and therefore high dissolution rate, one can decrease solubility through appropriate salt of derivative formation. Unfortunately, forms such as these do not meet the criterion of constant availability rate because their surface area decreases with time. Nevertheless, sustained drug release can be achieved by coating drug particles or granules with materials of varying thickness or by dispersing them in a polymeric matrix.
Principle:
If the dissolution process is diffusion layer controlled, where the rate of diffusion from the solid surface through an unstirred liquid film to the bulk solution is rate limiting, the flux J is given by:
J = -D (dc/dx) ---------- (1)
Where D is the diffusion coefficient and dc/dx is the concentration gradient from the solid surface to the bulk solution. The flux can also be defined as the flow rate to material (dm/dt) trough a unit area (A), thus one has the equation:
J = (1/A) dm/dt ---------- (2)
If the concentration gradient is linear and the thickness of the diffusion layer is h,
dc/dx = (Cb – Cs)/h ---------- (3)
Where Cs is the concentration at the solid surface and Cb is the concentration in the bulk solution. By combining the above equation, the flow rate of material is given by
dm/dt = -(DA/h)(Cb–Cs) = kA(Cs – Cb) ---------- (4)
Where k is the intrinsic dissolution rate constant.
The above equation predicts constant dissolution rate. If the surface area, diffusion co-efficient, diffusion layer thickness, and concentration difference are kept constant. However, as dissolution proceeds, all of the, parameters the surface area especially, may change.
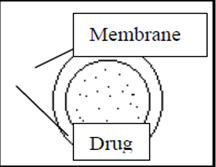
Figure 1.1: Dissolution control of drug release via thickness and dissolution rate of the membrane barrier coat.
Most suitable dosage forms for this mechanism is compressed tablets containing coated particles. E.g. Ethyl cellulose, Nylon, Acrylic resins. Release depends on drug solubility and pore structure membrane. Constant release resulted when GI fluid passes through barrier to dissolve drug.
b) Diffusion Controlled Release:
There are basically two types of diffusion-controlled systems, which have been developed over the past two decades: reservoir devices and matrix devices. In diffusion controlled extended release systems the transport by diffusion of dissolved drug in pores filled with gastric or intestinal juice or in a solid (normally polymer) phase is the release controlling process.
Depending on the part of the release unit in which the drug diffusion takes place, diffusion controlled release systems are divided into matrix systems (also referred to as monolithic systems) and reservoir systems.
In matrix systems diffusion occurs in pores located within the bulk of the release unit, and in reservoir systems diffusion takes place in a thin water-insoluble film or membrane, often about 5-20 μm thick, which surrounds the release unit. Diffusion through the membrane can occur in pores filled with fluid or in the solid phase that forms the membrane.
Drug is release from a diffusion controlled release unit in two steps-
1. The Liquid that surrounds the dosage from penetrates the release unit and dissolves the drug. A concentration gradient of dissolved drug is thus established between the interior and the exterior of the release unit.
2. The dissolved drug will diffuse in the pores of the release unit or the surrounding membrane and thus be released, or, alternatively, the dissolved drug will partition into the membrane surrounding the dose unit and diffuse in the membrane.
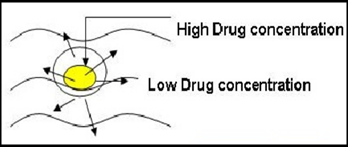
Figure 1.2: diffusion release pattern
A dissolution step is thus normally involved in the release process but the diffusion step is the rate-controlling step.
The rate at which diffusion will occur depends on four variables:
* The concentration gradients over the diffusion distance.
* The area.
* The distance over which diffusion occurs.
* The diffusion co-efficient of the drug in the diffusion medium.
Some of these variables are used to modulate the release rate in the formulation.
c) Osmotic pump system:
The rate of drug release in these products is determined by the constant inflow of water across semipermeable membrane into a reservoir, which contains an osmotic agent. The drug is either mixed with the agent or is located in a reservoir. The dosage form contains a small hole from which dissolved drug is pumped at a rate determined by the rate of entrance of water due to osmotic pressure.
The advantage of this type of product is that the constant release is unaltered by the environment of the gastrointestinal tract. The rate of release can modified by altering the osmotic agent and the size of the hole.
dm = Ak?ss ---------- (5)
dt h
Where, A =membrane area, k =membrane permeability, h =membrane thickness
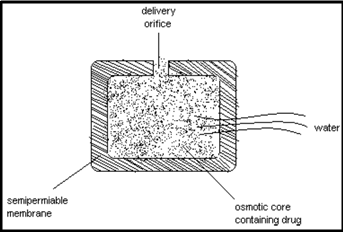
Figure 1.3: Osmotic pressure controlled by size of hole and concentration of osmotic agent in the core system.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
d) Erosion controlled release systems:
In erosion controlled extended release systems that rate of drug release is controlled by the erosion of a matrix in which the drug release is controlled by the erosion of a matrix in which the drug is dispersed. The matrix is normally a tablet, i.e. the matrix is formed by a tab letting operation, and the system can thus be described as a single unit system.
The erosion in its simplest form can be described as a continuous liberation of matrix material (both drug and excipients) from the surface of the tablet, i.e. surface erosion. The consequence will be a continuous reduction in tablet weight during the course of the release process.
Mechanism of drug release from a erosion based matrix tablet:
Drug release from an erosion system can thus be described in two steps.
1. Matrix material, in which the drug is dissolved or dispersed, is liberated from the surface of the tablet.
2. The drug is subsequently exposed to the gastrointestinal fluids and mixed with (if the drug is dissolved in the matrix) or dissolved in (if the drug is suspended in the matrix) the fluid.
The eroding matrix can be formed from different substances. One example is lipids or waxes, in which the drug is dispersed. Another example is polymers that gel in contact with water (Hydroxy ethyl cellulose). The gel will subsequently erode and release the drug dissolved or dispersed in the gel. Diffusion of the drug in the gel may occur in parallel.
e) Release is controlled by ion exchange:
Ion exchangers are water insoluble resinous materials containing salt forming anionic or cationic groups. While manufacturing, the drug solution is mixed with resin and dried to form beads which are tableted. The drug release depends upon high concentration of charged ions in gastro intestinal tract where, the drug molecules are exchanged and diffused out of the resin into the surrounding fluid. This mechanism relies upon the ionic environment of resin and not pH or enzyme on absorption site.
1.2 EXTENDED RELEASE SOLID ORAL DOSAGE FORMS7
Extended release (ER) dosage form is one of the drug products categorized under the term modified release dosage forms (FDA, 1997). It refers to products, which are formulated to make the drug available over an extended period after ingestion; thus, it allows a reduction in dosing frequency compared to a conventional type i.e. immediate release (IR) dosage form. Several advantages of ER products over IR ones have long been recognized. ER solid oral dosage forms can be classified into two broad groups:
(i) Single unit dosage forms (e.g. tablets) and
(ii) Multiple unit dosage forms or multiparticulate pellet systems.
The systems can be further subdivided into two concepts regarding to the design of dosage forms:
(i) Matrix systems and
(ii) Reservoir systems.
1.2.1 SINGLE UNIT DOSAGE FORMS
Matrix systems
Matrix or monolithic devices consist of drug dispersed homogenously throughout a continuous phase of polymer or lipid. The devices can be prepared either by the compression of a polymer/drug mixture or by the dissolution or melting, resulted in the molecularly dispersed drug. The drug transport often results from a combination of several mechanisms included dissolution, diffusion, swelling and erosion.
a. Water-soluble matrix formers
Water-soluble or hydrophilic matrices are a well known type of ER oral dosage forms. While hydroxypropyl methylcellulose (HPMC) is the most important hydrophilic carrier material, several others are also available; including
(i) Cellulose derivatives: hydroxypropyl cellulose (HPC), carboxymethylcellulose sodium (NaCMC),
(ii) Natural polymers: sodium alginate, carrageenan, chitosan
(iii) Synthetic polymers: polymerized acrylic acid (Carbopol), polyvinyl alcohol (PVA), polyethylene oxide (PEO). It has been suggested, however, that the term ‘swellable matrices’ is more appropriate as it better explains the characteristic of the systems.
b. Water-insoluble matrix formers
Water-insoluble carrier materials include
(i) Lipid-base excipients: white wax, carnauba wax, glycerylmonostearate, hydrogenated vegetable oil, paraffin and
(ii) Polymer-based excipients: ethylcellulose (EC), cellulose acetate. In comparison to the hydrophilic matrices, the system has a greater physical stability, resulting in the less variable drug release and the lower incidence of ‘dose dumping’ in presence of food.
Reservoir systems
Reservoir systems are characterized by a drug-containing core surrounded by release-rate controlling polymer(s). The mechanism of the drug transport across the polymeric membrane has been extensively described by Lecomte (2004).
a. Coated tablets
An example of technology for ER coated tablet is MODAS (Multiporous Oral Drug Absorption System; Elan Corporation, Ireland). The tablet core consists of the mixture of active drug and other excipients, subsequently coated with a solution of water-insoluble polymers and water-soluble excipients. Upon exposure to aqueous media, the surrounded coating is transformed into a semi-permeable membrane through which the drug diffuses in a rate-limiting manner.
b. Osmotic pump systems
Osmotic device is a special type of the reservoir systems, where the release rate of the drug is controlled dynamically by an incorporated osmotic agent in the active drug core. The rigid surrounding semi-permeable membrane consists for example of cellulose acetate. The drug is released through a defined, laser drilled delivery orifice in the membrane.
1.2.2 MULTIPARTICULATE PELLET SYSTEMS
Several advantages of multiparticulate systems over the single unit ones have been well documented. Following a proper preparation method, the ER pellets are either filled into a capsule or are compressed into a tablet.
Matrix systems
The matrix type of multiparticulate systems can be prepared by several techniques such as extrusion/spheronisation, spherical crystal agglomeration and melt-solidification. Although, the production of multiparticulate matrix systems is considered to be easier than that of the reservoir systems, their extent of retardation is limited because of pellet geometry.
Reservoir systems
Coated pellets as a mean to control drug delivery are widely used in the pharmaceutical industry, although the development and optimisation of the systems are rather complex. Numerous aspects of the system performance have been investigated, for instance, the influence of formulation and coating technique, the effect of drug solubility and core material, the use of polymer blends, in vitro/in vivo evaluation and the influence of release medium.
1.3 TABLET8,9
Tablets are solid dosage form containing ingredients with or without filler material. Tablets are oral solid dosage form of medicaments with or without suitable diluents and prepared either by molding or compression. They are solid, flat or biconvex disc in shape. They vary greatly in shape, size and weight which depend upon amount of medicament used and mode of administration. They also vary in hardness, thickness, disintegration and dissolution characteristics and in other aspects depending upon their intended use and method of manufacture. Tablets are the most widely used solid dosage form of medicament. Because of their advantages their popularity is continuously day by day.
Advantages of tablet dosage form:
* Tablet is intact dosage form and offers the best capabilities of all oral dosage forms for accuracy in size and content of the lowest variability.
* Tablet dosage form which is the lowest cost of manufacture (if it is calculated per dose).
* Tablets is an oral dosage form of the lightest, most compact, easiest and most inexpensive way to packed and shipped.
* The product identification on the tablets the easiest and inexpensive, requiring no additional work steps when using the printer surface that monogram or arising accessories.
* Tablet can be used as a product of specific release profiles, such as the release in the intestine or slow release products.
* A tablet is an oral dosage form of the most easy to be produced in bulk (large scale).
* Tablets are easy to use, handle and carry by the patient.
* Tablets provide prolonged stability to medicament.
* Tablets are provides a sealed covering which protects the tablets from atmospheric conditions like air, moisture and light etc.
* The unpleasant taste and odour of medicament can be easily masked by sugar coating.
* Whenever a fractional dose is required, tablets are divided into halves and quarters by drawing lines of tablet.
* Tablets provide administration of even minute dose of drug in an accurate amount.
* Tablets are formulated as a special release of products such as enteric or delayed release products.
Disadvantages of Tablet Dosage Form:
* Some drugs cannot be compressed into solid and compact, depending on its amorphous state, flocculation, or low density.
* Drugs moistened difficult, slow dissolves, moderate or high dose, high optimum absorption via the gastrointestinal tract or any combination of the properties above, it would be difficult or impossible to be formulated and fabricated in the form of tablets that produce sufficient drug bioavaibility.
* Medicine that tastes bitter, a drug with the smell was terrible and cannot be eliminated, or drugs that are sensitive to oxygen or air humidity needs to encapsulation or compression cloaking before (if possible) or require coating first. In this case, the capsule is a cheaper way out.
Types and classes of tablets:
a) Oral tablet for ingestion
* Compressed tablet
* Multiple compressed tablets
* Delayed action tablet
* Modified release tablet
* Sugar coated tablets
b) Tablet used in oral cavity
* Buccal tablets
* Sublingual tablets
* Troches and lozenges
* Dental cones
* Film coated tablets
* Chewable tablets
* Targeted tablet
c) Tablet administered by other routes
* Implantation tablets
* Vaginal tablets
d) Tablets used to prepare solution
* Effervescent tablets
* Dispensing tablets
* Hypodermic tablets
* Tablet triturates
Modified release tablet:
The main aim behind formulation of this dosage form is to release the medicament slowly for long time duration after administration of a single tablet. More over, these type of formulations are generally used to target the site specific releases.
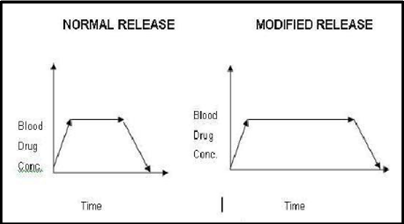
Figure 1.4: Graphical comparison of blood concentration v/s time
A widespread use of this type of tablet is seen in present scenario, as well as many researchers have concentrated their attention in this direction. This is mainly because of improvement in patient’s compliance as the dosage frequency is reduced, patient can take an undisturbed sleep at night, it’s also beneficial for psychiatric patients who forget to take their tablets regularly and the dose related side effects and toxicities are reduced. Any adjuvant that can alter water uptake rate, swelling, and gelling characteristics of matrixing agents can alter the release rate of API example like electrolytes in HPMC matrix tablet.
It’s also possible to achieve pulsed drug release. Weakly basic drugs exhibit good solubility at low pH while less soluble at high pH conditions, which can result in incomplete drug release for sustained release formulations. The drug release can be modified by providing suitable micro environmental pH in the tablet e.g., acidic polymer, succinic acid, etc. Similarly, inclusion of alkaline polymers results in desirable drug release of acidic drugs. On the other hand, formulation of this type of dosage form presents challenge for the formulator: increases the cost of manufacturing, chances of burst drug release and drop in drug release rate in terminal phase and thus incomplete release on API. In case of accidental poisoning, the doctor has to deal with special treatment problems. Due to large size, patient may feel difficulties in swallowing as the matrixing agent to drug ratio is high. Classic approaches are usually based on adaptation of either film coated or multiparticulate technologies or those involving slow release matrices.
1.4 CAPSULE10,11
The word ‘capsule’ in the English language is derived from the Latin word ‘capsula’, which means a small box or container. In more recent times, capsule has been used primarily to describe a solid oral dosage form, which consists of a container, usually made of gelatin, filled with a medicinal substance. There are many forms of capsules and they can be divided into two main categories, which in current English usage are described by the adjectives ‘hard’ and ‘soft’. The ‘hard capsule’ consists of two separate parts, each a semi-closed cylinder in shape. One part, the ‘cap’, has a slightly larger diameter than the other, which is called the ‘body’ and is longer. The cap fits closely over the body to form a sealed unit.
Figure 1.5: Self-lock capsule
Capsule is a solid particle which has a size of 0.1 to 10,000 μ. According to the pharmacopoeia of Indonesia, the capsule is a solid dosage of the drug in hard or soft shell that can be dissolved. Shells are generally made of gelatin, can also be made from starch or other suitable material.
Gelatin is the commercial protein derived from the native protein collagen, which is present in animal skin and bone, and the term ‘gelatin’ originates from the Latin ‘gelatus’, meaning stiff or frozen. Gelatin has all the properties required to meet the technical needs of the pharmaceutical capsule industry. These include solubility, solution viscosity and thermally reversible gelation properties in aqueous solution. It produces strong, clear, flexible, high-gloss films, which dissolve readily under the conditions existing in the stomach. Furthermore, current scientific evidence indicates that gelatin is a safe raw material.
Advantages of Capsule dosage form:
* Hard-gelatin capsules suitable for extemporaneous compounding so that the dose and combination of ingredients may vary depending on the patient's needs
* Stable than liquid dosage forms
* Can cover up the taste and smell unpleasant medicine
* Liquid preparations can be made with a certain concentration
* Used for depot capsules and enteric coated capsule
* Capsules, because of their elongated shape, are easy to swallow, which is one reason for the number of capsule-shaped tablets manufactured today.
* Biggest formulation advantage of capsules is that there is less need for additional excipients.
* Since capsules are tasteless, they effectively mask any unpleasant taste or odor of their contents.
* They offer rapid release characteristics, due to the rapid dissolution rate of the capsules.
* The use of hard capsules is also a common feature in clinical trials, as the filling of tablets or even capsules themselves will blind the dosage forms studied.
* Controlled release can be achieved using capsules. Dry powder mixtures, granules, pellets and tablets can be filled into hard capsules. Moreover combination of two or three types (i.e. dry powder mixtures, tablets or pellets) also can be put into capsules.
Disadvantages of Capsule:
* Not suitable for very soluble ingredients such as KCl, CaCl2, KBR, NH4Br. When the capsule is broken contact with the wall of the stomach, then the solution will be concentrated so that irritate the stomach and the stomach becomes tense.
* Can not be used for materials that are very efflorescent or deliquescent. Efflorescent material make capsule become soft while deliquescent material causing the capsule to become brittle and easily broken.
* The bitter-medicine will cause vomiting and corrosive which are difficult to overcome
* It took a relatively long compounding
Type of Capsules
There are various forms of capsule, including:
1. Based on consistency:
* Hard-capsule
* Soft-capsule
2. Based on how to use:
* Per Oral
* Per-rectal
* Per vaginal
* Topical
3. Based on purpose of use:
* For animals
* For human
Hard Gelatin Capsule
Hard gelatine capsules can be filled with a large variety of materials of different physicochemical properties (i.e. dry solids, semisolids, non-aqueous liquids, etc), while soft gelatin capsule are generally used to contain liquid and semisolid materials.
Specialist capsules have been made to meet the demands of certain applications, e.g. gastro-resistant capsules, modified-release capsules, self-locking capsules, capsules for liquid filling, capsules for administration to animals and capsules used for certain clinical trials. For current applications there are certain design features that all hard capsules must possess, viz. a feature to hold the empty capsule shells together, a self-locking feature, an air venting system and a feature to allow accurate rejoining after filling.
Hard gelatin capsule shell consisting of:
1. Basic ingredients: Gelatin, Sugar, Water
2. Other ingredients: Dyes, preservatives (eg SO2), Blur agent (eg TiO2), flavoring agent
The size and capacity of hard gelatin capsule shell :
1. For human: 000, 00, 0, 1, 2, 3, 4, 5
2. For animals: 10, 11, 12
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
1.5 ANTIBIOTIC12,13,14
An antibacterial is a compound or substance that kills or slows down the growth of bacteria.
The word antibiotic comes from the Greek anti meaning 'against' and bios meaning 'life' (a bacterium is a life form). Antibiotics are also known as antibacterials, and they are drugs used to treat infections caused by bacteria.
A chemical substance derivable from a microorganism orproduced by chemical synthesis that kills or inhibitsmicroorganisms and cures infections known as antibiotic.
A broad-spectrum antibiotic can be used to treat a wide range of infections. A narrow-spectrum antibiotic is only effective against a few types of bacteria.
Antibiotics are drugs used to kill or harm organisms such as bacteria, viruses, funguses and protozoon in living organisms. Since their discovery in the 1930s, antibiotics have made it possible to cure diseases caused by bacteria such as pneumonia, tuberculosis and meningitis- saving the lives of millions of people around the world. Some antibiotics are produced from live organisms such as bacteria and funguses. Other antibiotics are totally or partially produced synthetically.
Antibiotics act via two mechanisms: they kill the microorganisms (bactericide action) and prevent them from reproducing (bacteriostatic action).
Classification of antibiotic
1. β-Lactam antibiotics
Examples: penicillins (e.g. amoxicillin),
2. Cephalosporins, carbapenems, monobactams, etc.
3. Tetracyclines
Example: tetracycline
4. Macrolide antibiotics
Example: erythromycin
5. Aminoglycosides
Examples: Gentamicin, Tobramycin, Amikacin
6. Quinolones
Example: Ciprofloxacin (a fluoroquinolone)
7. Cyclic peptides
Examples: Vancomycin, Streptogramins, Polymyxins
8. Lincosamides
Example: clindamycin
9. Oxazolidinoes
Example: Linezolid (Zyvox)
10. Sulfa antibiotics
Example: sulfisoxazole
Antibiotics are among the most frequently prescribed medications in modern medicine. Antibiotics cure disease by killing or injuring bacteria. The first antibiotic was penicillin, discovered accidentally from a mold culture. Antibiotics can help treat infections caused by bacteria but not by viruses.
Amoxicillin, an acid stable, semi?synthetic drug belongs to a class of antibiotics called the Penicillins (B?lactam antibiotics).
Subsequent need to adjust antimicrobial therapy in light of the laboratory results:15
Since different organisms vary in their susceptibility to antimicrobial agents, it is imperative that we have some means for determining the antimicrobial susceptibility of the infecting organism(s). Once the pathogen has been isolated, it can be subjected to susceptibility testing.
The commonly used disc-diffusion method is relatively simple to perform and is the most widely employed method. It provides semi quantitative or qualitative data about the susceptibility of a given organism to a given agent. The qualitative assessment of susceptibility is generally categorised as sensitive or resistant; however, some laboratories also report an intermediate category.
Quantitative data are also provided by methods that incorporate serial dilutions of antimicrobials in agar-containing or broth culture media. The lowest concentration of the antimicrobial agent which inhibits visible growth after an18 - 24 hour incubation period is known as the minimal inhibitory concentration (MIC). The minimum bactericidal concentration (MBC) is determined in broth dilution tests by subculturing samples without visible growth; this is based on 99.9% killing after 18 to 24 hours of incubation.
Testing the ability of the cultured pathogen to grow or not at a critical concentration (chosen to distinguish between sensitive and resistant bacteria), is a modification known as "breakpoint" testing. A recently described modification of the classical MIC test, the E-test, uses diffusion of a continuous concentration gradient of an antimicrobial agent from a plastic strip into an agar medium to yield quantitative measurements of antimicrobial susceptibility.
Monitoring Therapeutic response:15
In many patients, it is possible to monitor the therapeutic response on clinical grounds alone. Thus the subsidence of fever, the return of well-being, and the disappearance of both local and systemic signs of infection in the patient, all signify an appropriate response. No further formal monitoring is necessary in most cases.
An apparent failure to respond clinically may be due to either ineffectiveness of antimicrobial agent(s) (due to resistance or inappropriate route of administration) or to other reasons e.g. a localised infection that requires surgical drainage, or a superinfection etc. Careful reassessment is recommended when considering changes of antimicrobial therapy.
In certain situations, measurement of antimicrobial activity may be useful in predicting clinical response, e.g. determination of serum bactericidal activity (Schlichter test) in cases of infective endocarditis.
Pharmacokinetic properties of antibiotics:15
Knowledge of the pharmacodynamic and kinetic properties of antibiotics is imperative in choosing the correct antibiotic and correct dose. In order for antibiotics to exert their bactericidal or bacteriostatic activity, a few important principles pertain:
1. Microbiological activity - antibiotic must bind to a specific binding site (e.g. ribosome or penicillin binding protein).
2. Concentration of the antibiotic at the site of the infection is important (the higher the concentration the more binding sites are occupied on/in the bacterial cells).
3. The antibiotics also have to remain on these binding sites for a sufficient period of time.
4. Minimum inhibitory concentration (MIC): This concentration represents the minimum amount of drug with which the bacteria have to come into contact, in order for the antibiotic towork.
1.6 PHARYNGITIS/TONSILITIS16,17
Pharyngitis and tonsillitis are defined as acute inflammations involving the posterior pharynx and the tonsillar pillars. The most common bacterial cause of pharyngitis and tonsillitis is group A beta-hemolytic streptococci. For regulatory purposes, it has been customary to request the actual species be identified (i.e., Streptococcus pyogenes).
Pharyngitis:
Pharyngitisis an inflammationof the throator pharynx, In most cases it is quite painful, and is the most common cause of a sore throat. Pharyngitis can result in very large tonsilswhich cause trouble swallowing and breathing. Bacteria responsible for pharyngitis:-Streptococcus spp.
Tonsillitis:
Tonsillitisis an inflammationof the tonsilsmost commonly caused by viral or bacterial infection. Symptoms of tonsillitis include sore throatand fever.
Signs and symptoms characteristic of pharyngitis/tonsillitis should include the following:
* A sore and scratchy throat, pain on swallowing (odynophagia), temperature, chills and/or fever.
* The pharyngeal mucosa should be erythematous to fiery red, and a thick exudate should cover the pharynx and tonsillar area.
* Uvular edema may be noted.
* Cervical adenopathy should be present and commented on.
* A white count over 12,000 may be present.
* Strains of S. pyogenesthat elaborate erythrogenic toxin may cause a scarlet fever rash of the face and skin folds, red tongue and prominent papillae (strawberry tongue).
* sore throat
* fever - either low grade or high
* headache
* decrease in appetite
* not feeling well
* nausea
* vomiting
* stomach aches
* painful swallowing
* visual redness or drainage in the throat
Objective Data:18
The following assessment is suggested, and any or all of these findings may be noted:
* Temperature:
The temperature is usually >100.5 degrees F in streptococcal infections and is usually <100.5 degrees F in viral cases.
* Skin:
Inspect for a rash. Occasionally a diffuse, erythematous rash with petechiae, which starts on the neck and extends downward, accompanies streptococcal infections.
* Oropharynx:
Inspect for redness, inflammation and exudates of the pharynx, uvula and tonsils. Exudate is more common with streptococcal infections.
* Neck:
Palpate for lymphadenopathy. Swollen and tender anterior cervical nodes are more often seen in streptococcal infections.
* Ears:
Perform an otoscopic examination. The examination is usually normal in isolated tonsillopharyngitis.
Pharyngitis and tonsillitis diagnosis:19
In most cases, it is hard to distinguish between a viral sore throat and a strep throat based on physical examination. It is important, though, to know if the sore throat is caused by Group A Beta Hemolytic Streptococci , as this requires antibiotic treatment to help prevent the complications that can occur with these bacteria.
As a result, most people, when they have the above symptoms, will receive a strep test and throat culture to determine if it is an infection caused by Group A Beta Hemolytic Streptococci. This usually involves a quick throat swab in the physician's office.
Quick tests, called rapid strep tests, may be performed. This may also immediately become positive for Group A Beta Hemolytic Streptococciand antibiotics will be started. If it is negative, part of the throat swab will be kept for a throat culture. This will further identify, in two to three days, if there is any Group A Beta Hemolytic Streptococcipresent. Physician will decide the treatment plan based on the findings.
Treatment for pharyngitis and tonsillitis:19
Specific treatment for pharyngitis and tonsillitis will be determined by your physician based on:
* Your age, overall health, and medical history
* Extent of the condition
* Cause of the condition
* Your tolerance for specific medications, procedures, or therapies
* Expectations for the course of the condition
* Your opinion or preference
If bacteria are not the cause of the infection, then the treatment is usually directed more for comfort. Antibiotics will not help treat viral sore throats. Treatment may include:
* Acetaminophen (for pain)
* Increased fluid intake
* Throat lozenges
* Antibiotics (if the cause of the infection is bacterial, not viral)
1.7 MICROBIOLOGY
Various study reports showed that amoxicillin was effective against variety of micro?organisms with MIC ranges 0.06 μg/ml?4 μg/ml for most of the micro?organisms. The absolute time unbound amoxicillin concentrations remained > MIC value of 0.06 μg/ml was ≈ 13 hours in healthy subjects. For Escherichia coli, the kill rates were higher with amoxicillin than with ampicillin with exponential bactericidal response. With an antibiotic half?life of 1 hr, the amoxicillin first order inactivation rate was 3.544 hr?1and the viable cell half?life was 0.196 hr; the respective values for ampicillin were 2.341 hr?1and 0.296 hr.For Staphylococcus aureus, the rates of kill were similar with both agents, but, amoxicillin had a long bacteriostatic phase which was not seen with ampicillin. This led to a longer lasting antibacterial effect and reduction to a lower total count with amoxicillin.20
In treating a bacterial infection, the amoxicillin product is formulated to provide a concentration of amoxicillin in the plasma that is above the MIC of the bacterial pathogen for a period of time each day that is effective for treating the bacterial infection.21
Table 1.1: Minimum Inhibitory Concentration of amoxicillin of several clinically important micro-organisms.22
|
In vitro activity |
Average minimal inhibitory concentration (MIC) |
||
|---|---|---|---|
|
0.01-0.1 mcg/ml |
0.1-1 mcg/ml |
1-10 mcg/ml |
|
|
Gram-positive micro-organisms |
Str.β haemolyticus |
Staph. aureus(penicillinase negative) |
Str. Faecalis |
|
Gram-negative micro-organisms |
N. gonorrhoeae |
H. influenzae |
E. coli |
The amoxicillin is the antibiotic agent. The main objective of present work is to formulate modified release dosage form of amoxicillin wherein its release is modified over conventional release. Such modified release formulations have been found to improve the bactericidal effect of drug on Pharyngitis/Tonsilitis. The modified release dosage form has the advantage that the total dose of amoxicillin decreases over conventional dosage form. They increase the patient compliance. Hence in this work objective is to formulate modified release system for in order to check plasma concentration profile for 24 hrs. and try to achieve the same.
OBJECTIVES OF THE RESEARCH WORK
The aim of this work would be to design and evaluate modified release dosage forms containing antibiotic agent.
Broadly, the work would endeavour to achieve the following objectives:
1) To prepare different modified release dosage form of amoxicillin for the treatment of pharyngitis/tonsillitis.
2) To study the Preformulation factors such as melting point, drug-excipients interaction, angle of repose, carr’s index, drug property etc.
3) To characterize manufactured tablets for hardness, thickness, content uniformity, weight uniformity, dimensions, etc. and capsule for weight uniformity, content uniformity, appearance.
4) To study in vitro drug release study comparison of different dosage form like tablet, capsule.
5) To study in vitro drug release study of different dosage form with marketed product in US or with the ideal theoretical drug release profile.
6) To carry out short term accelerated stability studies on the most satisfactory formulation as per ICH guidelines.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RATIONALE FOR STUDY
* Bacterial regrowth occurs rapidly after this antibiotic concentration fall below bacterial Minimum Inhibitory Concentration (MIC). Therefore should prevent drug free interval between doses from being long enough for the bacterial pathogen to resume growth.
* Continuous administration of amoxicillin decreases the toxicity.
* Kinetics of bactericidal effect is slow and requires prolonged maintenance of effective concentration of drug.
* A smaller total antibiotic dose is required to achieve the same pharmacodynamic endpoint by continuous infusion in comparison to intermittent infusion.
* Elevation of beta lactum concentration demonstrates increased bacterial killing, only until finite points which tends to be about 4times the MIC. Further elevation is not increase bactericidal potency. It decreases potency.
* A direct correlation exists between times the beta lactum antibiotic concentration is maintained above therapeutics concentration, so continuous administration advantage.
* Tissue penetration of these drugs is not correlated with serum concentrations, i.e. elevation of serum drug concentration will not contribute much in cases where the pathogen is located intracellularly.
* Beta lactum antibiotics exhibit short half life values, which demand frequent drug administration. Therefore continuous administration is beneficial.23
* The concept of delivering the drugs after a well defined lag phase leads to the development of more than one pulse (multiple pulses) delivery systems. The multiple pulse delivery system offers advantages over biological resistance to antibiotics. Spore forming bacteria in the dormant phase are more prone to getting killed.24
RATIONALE FOR DRUG SELECTION
* It is broad spectrum antibiotic. So kill both gram negative and gram positive bacteria.
* Limited range of non β?lactam antibacterials was available; most had certain limitations in terms of toxicity.
- E.g.sulphonamides (rashes and renal toxicity); streptomycin and kanamycin (ototoxicity and nephrotoxicity); chloramphenicol (bone marrow aplasia); erythromycin (gastrointestinal side effects); tetracyclines (concentrate in developing bones and teeth) colistin (neuro and nephro?toxicity).
- A number of beta?lactams, penicillins: penicillin G and V (gastric acid labile), ampicillin, methicillin (nephrotoxicity) cephalosporins: cephaloridine and cephalothin (nephrotoxicity).
* It is better absorbed than ampicillin when given by mouth.
* Absorption is not affected by the presence of food in the stomach.
* Various study reports showed that amoxicillin was effective against variety of micro?organisms with MIC ranges 0.06 μg/ml?4 μg/ml for most of the micro?organisms ( eg: Staphylococcus aureus, H.influenza, S.pneumonia, S.pyogene i.e. responsible for tonsilitis/pharyngitis. )20
* Penicillin antibiotics need to have chemical structures added to increase their acid stability.
* It is better absorbed than ampicillin when given by mouth.
* The incidence of gastrointestinal, hepatic and haematological side effects is significantly higher for amoxicillin/clavulanic acid than amoxicillin alone.
* Amoxicillin/clavulanic acid seems to be associated with a higher risk of Stevens-Johnson syndrome, purpura and hepatitis than amoxicillin alone.
* Amoxicillin/clavulanic acid have low stability than amoxicillin alone.25
In view of these objectives extensive literature review was done and was reported in the next chapter “Literature Review”.
3.1 REVIEW OF DRUG
PHYSICOCHEMICAL PROPERTIES 26, 27, 28
Drug Name:-AMOXICILLIN TRIHYDRATE
Chemical formula:-C16H19N3O5S.3H2O
Chemical name:- (6R)-6-(a -4-hydroxyphenyl-D- glycylamino)penicillanic acid trihydrate.
Molecular Weight:-419.45
Chemical structure:
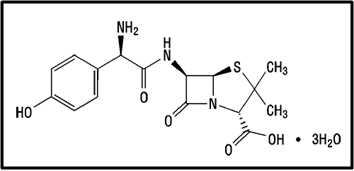
Description:- White or almost white, crystalline powder.A broad-spectrum semisynthetic antibiotic similar to ampicillin except that its resistance to gastric acid permits higher serum levels with oral administration.
Solubility:- Slightly soluble in water, in ethanol (95%) and in methanol; practically insoluble in chloroform, in ether and in fixed oils. It is soluble in dilute solutions of acids and of alkali hydroxides. (water solubility : 3430 mg/L)
Storage condition:- Store in tightly-closed containers in a cool place. Amoxicillin should be kept in the container it came in, tightly closed, and out of reach of children. Capsules and tablets of amoxicillin are crystalline in structure, so they should be stored at room temperature and away from excess heat and moisture (not in the bathroom). The liquid medication preferably should be kept in the refrigerator, but it may be stored at room temperature.
Melting range:-194 oC
Category:-Antibiotic
Heavy metals:-Not more than 20 ppm
Category:-Antibacterial
PHARMACODYNAMICS 29, 30, 31, 32
Amoxicillin is a moderate-spectrum antibiotic active against a wide range of Gram-positive, and a limited range of Gram-negative organisms. It is usually the drug of choice within the class because it is better absorbed, following oral administration, than other beta-lactam antibiotics.
Amoxicillin is stable in presence of gastric juices and it also produces less gastric disturbance and has the same antibacterial activity as ampicillin. It is a drug of choice in treatment of typhoid, meningitis, endocarditis, septicaemia, peritonitis and gonorrhoea. Another advantage of amoxicillin is that it penetrates equally well in to the purulent and mucoid sputum in distinction to ampicillin which does not cross the bronchial mucosa.
Amoxicillin is in the free acid form is a white crystalline powder sparingly soluble in water, It is stable in acid solution and thus can be given by mouth. It is well absorbed and produces high serum levels.
Peak serum concentrations are obtained within 60 to 90 minutes after drug administration. Animal studies demonstrated that amoxicillin is distributed evenly throughout the body tissues and is concentrated in the liver and kidneys. Small quantities enter the non infected cerebrospinal fluid. Administration of high doses results in proportionate increase in serum levels in patients with normal renal function. The drug is excreted in an active form in urine.
PHARMACOLOGY29, 30, 31, 32
Mechanism of action:-
Amoxicillin binds to penicillin-binding protein 1A (PBP-1A) located inside the bacterial cell well. Penicillins acylate the penicillin-sensitive transpeptidase C-terminal domain by opening the lactam ring. This inactivation of the enzyme prevents the formation of a cross-link of two linear peptidoglycan strands, inhibiting the third and last stage of bacterial cell wall synthesis. Cell lysis is then mediated by bacterial cell wall autolytic enzymes such as autolysins; it is possible that amoxicillin interferes with an autolysin inhibitor.
Bactericidal; inhibit bacterial cell wall synthesis. Action is dependent on the ability of penicillins to reach and bind penicillin-binding proteins (PBPs) located on the inner membrane of the bacterial cell wall.
The more rapid bactericidal activity is linked with a different effect on the growing cells. Thus while ampicillin and some other antibiotics such as cephalexin interfere primary with septation, resulting initially in elongated filamentous forms of gram negative bacteria, amoxicillin causes rapid interference with the cell wall leading to the formation of spheroplasts and lysis.
The cell wall of bacteria is essential for the normal growth and development. Peptidoglycan is a heteropolymeric component of the cell wall that provides rigid mechanical stability by virtue of its highly cross linked lattice work structure. In gram positive organism the cell wall is 50 to 100 molecules thick, while in gram negative micro organisms it is only 1 or 2 molecules thick. The biosynthesis of peptidoglycan involves about thirty bacterial enzymes and may be considered in three stages, which are as follows:
* The first stage involves the precursor formation which takes place in cytoplasm. The product, uridinediphosphate (UDP) accumulates in the synthesis of this compound is the addition of a dipeptide. D-alanyl-D alanine.
* During reactions of the second stage, UDP acetylmuramylpentapeptide and UDP acetylglucoseamine is linked to form a long polymer. To form this species, the sugar pentapeptide is first attached by a pyrophosphate bridge to a phospholipid in the cell membrane. The second sugar is then added, followed by the addition of five glycine residue as a branch of the heteropentapeptide. The molecule is then assumed to flip across the cell membrane such that the peptidoglycan precursor faces the periplasm. The completed unit is then cleaved from the membrane bound phospholipid.
* The final stage involves the completion of the cross link. This is accomplished by a transpeptidation reaction that occurs outside the cell membrane. The transpeptidase itself is membrane bound. The terminal glycine residue of the pentaglycine bridge is linked with the fourth residue of the pentapeptide (D-alanine) releasing the fifth residue (also D-alanine). It is this last step in peptidoglycan synthesis that is inhibited by the beta lactam antibiotics.
The lysis of bacteria that usually folloes their exposure to beta lactam antibiotics ultimately dependent on the cell wall autolytic enzymes i.e. autolysins. The relationship between inhibition of penicillin binding proteins (PBP) activity and activation of autolysins is unclear. Some evidence suggests that exposure of bacteria to beta lactam antibiotics results in the loss of an inhibitor of the autolysins.
Bata lactam antibiotics can interfere with cell wall synthesis only in growing cells, but presumably this antibiotics can bind to the transpeptidase and related enzymes even in resting cells thus inhibiting the terminal stages of cell wall synthesis if growth is subsequently resumed (Selwyn, 1980).
Indication33:-
For the treatment of infections of the ear, nose, and throat, the genitourinary tract, the skin and skin structure, and the lower respiratory tract due to susceptible (only b-lactamase-negative) strains of Streptococcus spp. (a- and b-hemolytic strains only), S. pneumoniae, Staphylococcus spp., H. influenzae, E. coli, P. mirabilis, or E. faecalis. Also for the treatment of acute, uncomplicated gonorrhea (ano-genital and urethral infections) due to N. gonorrhoeae (males and females).
· Infections of the ear, nose, and throat
Due to Streptococcus species. (α- and β-hemolytic isolates only), Streptococcus pneumoniae, Staphylococcus spp., or Haemophilusinfluenzae.
· Infections of the genitourinary tract
Due to Escherichia coli, Proteus mirabilis, or Enterococcus faecalis.
· Infections of the skin and skin structure
Due to Streptococcus spp. (α- and β-hemolytic isolates only), Staphylococcus spp., orE.coli.
· Infections of the lower respiratory tract
Due to Streptococcus spp. (α- and β-hemolytic isolates only), S. pneumoniae,Staphylococcus spp., or H. influenzae.
· Gonorrhea, acute uncomplicated (ano-genital and urethral infections in males and females) due to Neisseria gonorrhoeae.
· Triple therapy for Helicobacter pylori with clarithromycin and lansoprazole
· Dual therapy for H. pylori with lansoprazole
Contraindications33:-
Contraindicated in patients with known serious hypersensitivity to amoxicillin or to other drugs in the same class or patients who have demonstrated anaphylactic reactions to beta-lactams.
Drug Interactions33:-
* Probenecid
Probenecid decreases the renal tubular secretion of amoxicillin. Probenecid may result in increased and prolonged blood levels of amoxicillin. The clinical relevance of this finding has not been evaluated.
* Other Antibiotics
Chloramphenicol, macrolides, sulfonamides, and tetracyclines may interfere with the bactericidal effects of penicillin. This has been demonstrated in vitro; however, the clinical significance of this interaction is not well documented.
* Oral Contraceptives
As with other antibiotics, amoxicillin may affect the gut flora, leading to lower estrogen reabsorption and potentially resulting in reduced efficacy of combined oral estrogen/progesterone contraceptives.
Overdose33:-
In case of overdose, discontinue medication, treat symptomatically, and institute supportive measures as required. If the overdose is very recent and there is no contraindication, an attempt at emesis or other means of removal of drug from the stomach may be performed. A prospective study of 51 pediatric patients at a poison-control center suggested that overdosages of less than 250 mg/kg of amoxicillin are not associated with significant clinical symptoms and do not require gastric emptying.
Interstitial nephritis resulting in oliguric renal failure has been reported in a small number of patients after overdosage with amoxicillin.
Crystalluria, in some cases leading to renal failure, has also been reported after amoxicillin overdosage in adult and pediatric patients. In case of overdosage, adequate fluid intake and diuresis should be maintained to reduce the risk of amoxicillin crystalluria.
Renal impairment appears to be reversible with cessation of drug administration. High blood levels may occur more readily in patients with impaired renal function because of decreased renal clearance of amoxicillin. Amoxicillin may be removed from circulation by hemodialysis.
PHARMACOKINETICS29, 30, 31
Absorption:-
Rapidly absorbed after oral administration.
Distribution:-
Amoxicillin plasma protein binding is approximately 20%. The substance remains extracellular. The tissue concentrations depend on the circulation in those tissues and on the quantity of extracellular fluid. Amoxicillin diffuses adequately into the sputum, mucosa, bone tissue and aqueous humor of the eye to produce therapeutically active levels.
The concentrations in the bile are two to four times higher, or even higher than those in the blood. In the amniotic fluid and umbilical cord blood 25-30% of the mother's blood levels are attained. Amoxicillin diffuses poorly into the cerebrospinal fluid of patients with normal meninges. In inflamed meninges the concentrations are approximately 20% of those found in the blood.
Metabolism:-
Hepatic metabolism accounts for less than 30% of the biotransformation of most penicillins.
Elimination:-
Most of the amoxicillin is excreted unchanged in the urine; its excretion can be delayed by concurrent administration of probenecid.Amoxicillin is primarily eliminated via the kidneys, largely (ca. 80%) via tubular excretion, for the remainder (ca. 20%) via glomerular filtration.
Approximately 60% of an orally administered dose of amoxicillin is excreted in the urine within 6 to 8 hours. Detectable serum levels are observed up to 8 hours after an orally administered dose of amoxicillin.
Volume of distribution:- 0.3 L/kg
Half life:-61.3 min
pH:- 3.5-5.5
pKa:-9.48
Plasma Protein Binding:-In blood serum, amoxicillin is approximately 20% protein-bound
Dose:-The equivalent of 750 mg to 4.5 g of amoxycillin daily, in divided doses.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
3.1.1 DRUG PROFILE:
Table 3.1: Test, Specification and Result of Amoxicillin trihydrate as per IP.
|
Sr. No. |
TEST |
SPECIFICATION |
RESULT |
|
1 |
Description |
White or almost white, crystalline powder |
White crystalline powder |
|
2 |
Solubility |
Slightly soluble in water, methanol and ethanol (95%), soluble in dilute solutions of acids and of alkali-hydroxides. Practically insoluble in chloroform, in ether and in fixed oils. |
Slightly soluble in water, methanol and ethanol (95%), soluble in dilute solutions of acids and of alkali-hydroxides. Practically insoluble in chloroform, in ether and in fixed oils. |
|
3 |
Identification a) I.R absorption spectrum
b) By HPLC |
IR spectrum of sample as KBr pellet should be concordant with the spectrum obtained with working standard.
In the assay, the principal peak in the chromatogram obtained with the test solution corresponds to the peak in the chromatogram obtained with the reference solution. |
IR spectrum of sample as KBr pellet should be concordant with the spectrum obtained with working standard.
In the assay, the principal peak in the chromatogram obtained with the test solution corresponds to the peak in the chromatogram obtained with the reference solution. |
|
4 |
pH |
Between 3.5 and 5.5 |
4.42 |
|
5 |
Water content (by KF, % w/w) |
11.5 % to 14.5 % w/w |
13.14 |
|
6 |
Specific optical rotation (20% w/v solution in co2 free water |
Between +2900 and +3150 (on anhydrous basis) |
+303.5 |
|
7 |
Heavy metals |
Not more than 20 ppm |
Less than 20 |
|
8 |
Sulphated ash (%w/w) |
Not more than 1% w/w |
0.09 |
|
9 |
Assay (by HPLC) |
Not less than 95.0% and Not more than 100.5% w/w |
99.3 |
3.2 REVIEW OF EXCIPIENTS
EXCIPIENTS PROFILE34
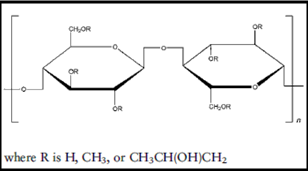
HYDROXY PROPYL METHYL CELLULOSE:
Synonym: Hypromellose, Methocel, Methylcellulose, HPMC
Chemical name: Cellulose, 2-hydrxy propyl methyl ether.
Chemical Formula: HPMC is a partially o-methylated and o-(2-hydroxypropylated) cellulose. It is available in several grades; vary in viscosity and extent of substitution.
Structural formula:
Molecular Weight: 10 000 – 1 500 000
Functional category: Tablet binder, suspending agent, coating agent, thickening agent, viscosity increasing agent.
Description:
HPMC is an odourless and tasteless, white or creamy with fibrous or granular powder.
Solubility:
Soluble in cold water forming a viscous colloidal solutions practically insoluble in chloroform, ethanol but soluble in mixtures of ethanol. Certain grades of HPMC are soluble in aqueous acetone solution.
Viscosity:
A wide range of viscosity types is commercially available. Aqueous solutions are most commonly prepared. Increasing concentration also produce more viscous solutions.
Stability and storage conditions: HPMC powder is stable material. Solutions are stable at PH 3-11. Aqueous solutions are comparatively enzyme resistant providing good viscosity stability during long-term storage. Hypromellose powder should be stored in a well-closed container.
Incompatibilities: It is incompatible with some oxidizing agent. Since it is nonionic, it will not complex with metallic salts and ionic organics to form insoluble precipitates.
Safety: It is generally regarded as a nontoxic and non-irritant material although excessive oral consumption may have a laxative effect.
Applications in pharmaceutical formulation:
· HPMC is widely used in oral, ophthalmic and topical formulations. Concentrations between 2% and 5% w/w may be used as a binder in either wet- or dry-granulation processes. High-viscosity grades may be used to retard the release of drugs from a matrix at levels of 10–80% w/w in tablets and capsules.
· HPMC is primarily used as tablet binder and as a matrix for use in extended release tablet formulation.
· Lower viscosity grades used in aqueous film coating solutions while higher viscosity grades are used with organic solvents.
· HPMC is also used as a suspending agent and thickening agents in topical formulations.
· HPMC at concentrations between 0.45%-1.0%w/w may be added as a thickening agent to vehicles for eye drops
ETHYL CELLULOSE
Synonyms: Ethocel, surelease, aquacoat ECD.
Structural formula:
Chemical Formula:Cellulose ethyl ether. A long chain polymer consisting of anhydroglucose units joined together by acetal linkages.
Description: Ethyl cellulose is a tasteless, free-flowing, white to light tan colored powder.
Functional Category: Coating agent, tablet binder, viscosity increasing agent.
Solubility: Practically insoluble in glycerin, propylene glycol, and water. It is freely soluble in chloroform, methyl acetate, tetrahydrofuran, and in mixtures of aromatic hydrocarbons with ethanol, methanol, and toluene.
Stability and Storage Conditions: Ethyl cellulose is a stable, slightly hydroscopic material. It is chemically resistant to alkalis, both dilute and concentrated, and to salt solutions, although it is more sensitive to acidic materials than cellulose esters. It is subject to oxidative degradation in the presence of sunlight or UV light at elevated temperatures.
The bulk material should be stored in a dry place, in a well closed container at the temperature between 7-32 °C.
Incompatibilities: It is incompatible with paraffin wax and microcrystalline wax.
Safety: It is widely used in oral and topical pharmaceutical formulations. It is also used in food products. It is generally regarded as a nontoxic, nonallergic and nonirritant material. Parenteral used may be harmful to the kidneys.
Applications:
· Ethyl cellulose is widely used in oral and topical pharmaceutical formulations.
· The main use of ethyl cellulose in oral formulations is as a hydrophobic coating agent for tablets and granules. Ethyl cellulose coatings are used to modify the release of a drug, to mask an unpleasant taste, or to improve the stability of a formulation; for example, where granules are coated with ethyl cellulose to inhibit oxidation.
· Modified release tablet formulations may also be produced using ethyl cellulose as a matrix former.
· Higher-viscosity ethyl cellulose grades tend to produce stronger and more durable films.
· An aqueous polymer dispersion (or latex) of ethylcellulose such as Aquacoat ECD (FMC Biopolymer) or Surelease (Colorcon) may also be used to produce ethyl cellulose films without the need for organic solvents.
· Drug release through ethyl cellulose-coated dosage forms can be controlled by diffusion through the film coating. This can be a slow process unless a large surface area (e.g. pellets or granules compared with tablets) is utilized. In those instances, aqueous ethyl cellulose dispersions are generally used to coat granules or pellets.
· Ethyl cellulose produces hard tablets with low friability, although they may demonstrate poor dissolution.
· In topical formulations, ethyl cellulose is used as a thickening agent in creams, lotions, or gels, provided an appropriate solvent is used. Ethyl cellulose has been studied as a stabilizer for emulsions.
EUDRAGIT
Nonproprietary Name: NF: Methacrylic acid copolymer; polymeric methacrylates.
Chemical name:Poly(methacrylic acid, methyl methacrylate) 1 : 1 (Eudragit L 100), Poly(methacrylic acid, methyl methacrylate) 1 : 2 (Eudragit S 100)
Functional Category: Film former, tablet binder
Synonyms: Eudragit, polymeric methacrylates.
Molecular Weight: ≥ 100, 000
Solubility: Type E, L and Type S – soluble in polar organic solvents, such as alcohols (ethanol, isopropanol), acetone, esters, chloroform, etc. Insoluble in water, petroleum ether, and saliva. Swells and dissolves in acidic media.
Type RL and RS – soluble in isopropanol and ethanol in combination with acetone or methylene chloride;also in methanol, chloroform, trichloroethylene, ethyl acetate, and glycolmonomethyl ether.
Eudragit L 100 Powder Soluble in intestinal fluid from pH 6, Eudragit S 100 Powder Soluble in intestinal fluid from pH 7
Safety: Acute toxicity studies have been performed in rats, rabbits, and dogs. No toxic effects were observed with Type L30D, E, L, S, RS, and RL at doses of dry lacquer substance ranging from 6-28 g/ kg body weight over a two- week period.
Applications:
- Binder – It has been used as a granulation binder in concentration between 5 to 20%.
- Film former – Eudragit E is used as film coating agent, soluble in gastric fluid. Eudragit RL and RS form water- insoluble film coat for delayed- release products.
PVP
Synonym: poly [1-(2-oxo-1-pyrrolidinyl)ethylene]; polyvidone; polyvinylpyrrolidone; 1-vinyl-2-pyrrolidinone polymer.
Chemical name: 1-Ethenyl-2-pyrrolidinone homopolymer
Molecular weight: 2500–3 000 000
Structural formula:
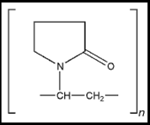
Functional Category: Disintegrant; dissolution aid; suspending agent; tablet binder.
Description: Povidone occurs as a fine, white to creamy-white colored, odourless or almost odourless, hygroscopic powder. Povidones with K-values equal to or lower than 30 are manufactured by spray-drying and occur as spheres. Povidone K-90 and higher K-value povidones are manufactured by drum drying and occur as plates.
Solubility: freely soluble in acids, chloroform, ethanol (95%), ketones, methanol, and water; practically insoluble in ether, hydrocarbons, and mineral oil. In water, the concentration of a solution is limited only by the viscosity of the resulting solution, which is a function of the K-value.
Viscosity (dynamic): the viscosity of aqueous povidone solutions depends on both the concentration and the molecular weight of the polymer employed.Dynamic viscosity of 5% w/v Povidone K-30 (Kollidon) solutions in ethanol (95%) is 3.4mPas and in propan-2-ol is 5.8mPas at 250C.
Applications:
· Intableting, povidone solutions are used as binders in wetgranulation processes.
· Povidone is used as a solubilizer in oral and parenteral formulations and has been shown to enhance dissolution of poorly soluble drugs from solid-dosage forms.
· Povidone solutions may also be used as coating agents.
· Povidone is additionally used as a suspending, stabilizing, or viscosity-increasing agent in a number of topical and oral suspensions and solutions.
· The solubility of a number of poorly soluble active drugs may be increased by mixing with povidone.
Table 3.2: Use of povidone
|
Use |
Concentration (%) |
|
Carrier for Drugs |
10-25 |
|
Dispersing agent |
Upto 5 |
|
Eye Drops |
2-10 |
|
Suspending Agent |
Upto 5 |
|
Tablet binder |
0.5-5 |
COLLOIDAL SILICON DIOXIDE:
Synonyms: Aerosil, colloidal silica, cab-o-sil, fumed silica; light anhydrous silicic acid; silicic anhydride
Chemical formula: Sio2
Functional category: Adsorbent, anticaking agent, glidant, suspending agent, viscosity improving agent.
Description: It is a light, loose, bluish-white colored, odorless, tasteless, nongritty amorphous powder.
Solubility: Practically insoluble In organic solvents, water and acids. It is soluble in hot solutions of alkali hydroxide.
Stability and storage conditions: Aerosil is hygroscopic but absorbs large quantities of water without liquefying. Aerosil should be stored in well-closed container.
Applications in pharmaceutical formulations:
· Colloidal silicon dioxide improves the flow properties of the dry powders. Aerosil also act as a thickening agent and suspending agent in gels and semisolid preparations.
· In aerosols Aerosil is used to promote particulate suspension, eliminate hard settling.
· It is also used as a tablet disintegrant.
· Aerosil is frequently added suppository formulations containing lipophilic excipients to increase viscosity, prevent sedimentation.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
MICROCRYSTALINE CELLULOSE:
Synonym: Avicel, celphere, emocel, cellulose gel, crystalline cellulose
Chemical name: Cellulose
Structural formula:
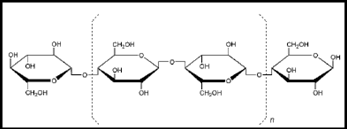
Functional category: Adsorbent, Suspending Agent, Diluent, Disintigrant.
Applications in pharmaceutical formulations:
· MCC is widely used in pharmaceutical formulation as a binder, diluent in tablet and capsule preparations.
· It also used as a disintegrant and lubricant.
Table 3.3: Uses of MCC
|
Use |
Concentration (%) |
|
Adsorbent |
20-90 |
|
Antiadherent |
5-20 |
|
Capsule binder/diluent |
20-90 |
|
Tablet disintegrant |
5-15 |
|
Tablet binder/diluent |
20-90 |
Description:
It is a white, odorless, tasteless, crystalline powder composed of porous particles. It is commercially available different particle sizes and moisture grades that have different properties and applications
Solubility:
MCC is slightly soluble in 5%sodium hydroxide solution. Practically insoluble in water, dilute acids and most organic solvents.
Stability and storage conditions:
MCC is a stable though hygroscopic material. The bulk material should be stored in a well-closed container in a cool, dry place.
TRIETHYL CITRATE:
Synonyms:Citric acid, ethyl ester
Chemical Name:2-Hydroxy-1,2,3-propanetricarboxylic acid, triethyl ester
Structural Formula:
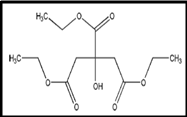
Functional Category: Plasticizer.
Description: Triethyl citrate is a clear, odorless, practically colorless, oilyliquid.
Solubility: soluble 1 in 125 of peanut oil, 1 in 15 of water. Miscible with ethanol (95%), acetone, and propan-2-ol.
Viscosity (dynamic): 35.2 mPa s (35.2 cP) at 250C
Stability and Storage Conditions: Triethyl citrate should be stored in a closed container in a cool, dry location. When stored in accordance with these conditions, triethyl citrate is a stable product.
Incompatibilities: Triethyl citrate is incompatible with strong alkalis andoxidizing materials.
Safety: Triethyl citrate is used in oral pharmaceutical formulations andas a direct food additive. It is generally regarded as a nontoxicand nonirritant material.
Applications:
· Triethyl citrate and the related esters acetyltriethyl citrate, tributyl citrate, and acetyltributyl are used to plasticize polymers in formulated pharmaceutical coatings.
· The coating applications include capsules, tablets, beads, and granules for taste masking, immediate release, sustainedrelease, and enteric formulations.
· Triethyl citrate is also used as a direct food additive for flavoring, for solvency, and as a surface active agent.
TRIACETIN:
Synonyms: Glycerol triacetate; glyceryl triacetate; triacetylglycerine.
Chemical Name:1,2,3-Propanetriol triacetate
Molecular weight:218.21
Structural formula:
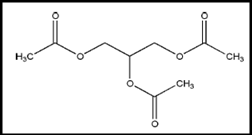
Functional category: Humectant; plasticizer; solvent.
Description: Triacetin is a colorless, viscous liquid with a slightly fatty odor.
Stability and Storage Conditions:Triacetin is stable and should be stored in a well-closed,nonmetallic container, in a cool, dry place.
Incompatibilities:Triacetin is incompatible with metals and may react withoxidizing agents. Triacetin may destroy rayon fabric.
Applications:
· Triacetin is mainly used as a hydrophilic plasticizer in both aqueous and solvent-based polymeric coating of capsules, tablets, beads, and granules; typical concentrations used are 10–35% w/w.
· Triacetin is used in cosmetics, perfumery, and foods as a solvent and as a fixative in the formulation of perfumes and flavors.
TITANIUM DIOXIDE:
Synonyms:Anatase titanium dioxide; brookite titanium dioxide; rutile titanium dioxide; titanic anhydride.
Molecular Weight:79.88
Structural Formula:TiO2
Functional Category:Coating agent; opacifier; pigment.
Description: White, amorphous, odorless, and tasteless nonhygroscopicpowder. Although the average particle size of titanium dioxidepowder is less than 1 mm, commercial titanium dioxidegenerally occurs as aggregated particles of approximately100 mm diameter.Titanium dioxide may occur in several different crystallineforms: rutile; anatase; and brookite. Of these, rutile and anataseare the only forms of commercial importance.
Solubility: practically insoluble in dilute sulfuric acid, hydrochloric acid, nitric acid, organic solvents, and water. Soluble in hydrofluoric acid and hot concentrated sulfuric acid.
Stability and Storage Conditions: Titanium dioxide is extremely stable at high temperatures. Thisis due to the strong bond between the tetravalent titanium ionand the bivalent oxygen ions. Titanium dioxide should be stored in a well-closed container, protected from light, in a cool, dry place.
Applications:
· Titanium dioxide is widely used in confectionery, cosmetics, and foods, in the plastics industry, and in topical and oral pharmaceutical formulations as a white pigment.
· Owing to its high refractive index, titanium dioxide has light-scattering properties that may be exploited in its use as a white pigment and opacifier.
· Titanium dioxide is also used in dermatological preparations and cosmetics, such as sunscreens.
MAGNESIUM STEARATE
Synonyms: Magnesium octadecanoate; octadecanoic acid, magnesium salt, stearic acid, magnesium salt.
Chemical name:Octadecanoic acid magnesium salt
Empirical Formula: C36H70MgO4
Molecular weight: 591.34
Structural Formula: CH3(CH2)16COO)2Mg
Functional Category: Tablet and capsule lubricant.
Description: Magnesium stearate is a fine, white, precipitated or milled, impalpable powder of low bulk density, having a faint odor of stearic acid and a characteristic taste. The powder is greasy to the touch and readily adheres to the skin.
Solubility: practically insoluble in ethanol, ethanol (95%), ether and water; slightly soluble in warm benzene and warm ethanol (95%).
Stability and storage conditions: Magnesium stearate is stable and should be stored in a well closed container in a cool, dry place.
Incompatibilities: Incompatible with strong acids, alkalis, and iron salts. Avoid mixing with strong oxidizing materials. Magnesium stearate cannot be used in products containing aspirin, some vitamins, and most alkaloidal salt.
Applications: Magnesium stearate is widely used in cosmetics, foods, and pharmaceutical formulations. It is primarily used as a lubricant in capsule and tablet manufacture at concentrations between 0.25% and 5.0% w/w. It is also used in barrier creams.
TALC
Synonyms:Hydrous magnesium calcium silicate; hydrous magnesium silicate; magnesium hydrogen metasilicate; MagsilOsmanthus.
Functional Category: Anticaking agent; glidant; tablet and capsule diluents and lubricant.
Empirical Formula: Talc is a purified, hydrated, magnesium silicate, approximating to the formula Mg6 (Si2o5)4 (OH)4. It contains small, variable amounts of aluminum silicate and iron.
Description: Talc is a very fine, white to grayish- white, odorless, impalpable, unctuous, crystalline powder. It adheres readily to the skin and is soft to the touch and free from grittiness.
Stability and storage conditions: Talc is a stable material and may be sterilized by heating at 160oC for not less than 1 hour. It may also be sterilized by exposure to ethylene oxide or gamma irradiation.Talc should be stored in a well-closed container in a cool, dry place.
Applications: Talc was once widely used in oral solid dosage formulations as a lubricant and diluent. It is widely used as dissolution retardant in the development of controlled-release products.
3.3 REVIEW OF DOSAGE FORM:
1. MiddleBrook Pharmaceuticals, Inc. has developed a delivery technology called PULSYS, which enables pulsatile delivery or delivery in rapid bursts of certain drugs. The technology provides the prolonged release and absorption of a drug. The company’s PULSYS product MOXATAG (amoxicillin extended-release) tablets, 775 mg are used for the treatment of pharyngitis/tonsillitis secondary to Streptococcus pyogenes, commonly known as strep throat, for adults and pediatric patients age 12 and older.35
2. K. Mallikarjuna Rao et al(2011) prepared floating microspheres of amoxycillin trihydrate agaistH.pylorias model drug for prolongation of gastric residence time. The microspheres were prepared by the Non Aqueous solvent diffusion method using polymers hydroxypropylmethyl cellulose and ethyl cellulose.In vitro drug release studies were performed, and drug release kinetics was evaluated using the linear regression method. The prepared microspheres exhibited prolonged drug release (10 h).36
3. JayeshParmaret al(2010) worked onMultiparticulate systems for oral extended release have been successfully developed and marketed due to their unique manufacturer and patient benefits that are realized. The formulation advantages are release profile modulation and combination therapy, and clinical benefits are ease of swallowing (paediatric and geriatric focus), consistent release profiles and minimal risk of dose dumping. This article will review the critical formulation aspects of multiparticulate core manufacturing considerations with emphasis on barrier membrane coating using ethylcellulose for oral extended release applications.37
4. Amnon Hoffman et al(1998) developed an oral sustained-release formulation for amoxicillin that maximize the duration of active drug concentration, thus increasing the dosing interval. Due to short biological half-life and limited ‘absorption window’ (confined to the small intestine) with poor colonic absorption, the matrix tablet formulation, composed of hydrophilic (hydroxypropyl methyl-cellulose) polymer. The results suggest that in order to achieve a twice daily dosing regimen that will provide therapeutic concentrations for the whole 12 h dosing intervals, a larger dose of the new formulation should be given (e.g. 750 mg or even 1g twice daily).23
5. Singhal M. et al(2011) developed extended release antipsychotic oral solid dosage formulation which exhibited better stability, high production feasibility and excellent patient acceptability compared to existing formulation. In the present study, Perphenazine depot tablets were formulated which included aqueous granulation as well as aqueous sustained release coating using different approaches in which different grades of Eudragits.38
6. Shiva Kumar Yellankiet al (2010) developed a mucoadhesive microsphere which improved drug delivery systems to gastro intestinal tract for treatment of Helicobacter pylori induced peptic and duodenal ulcers, In an effort to augment the anti-Helicobacter pylori effect of Amoxicillin trihydrate mucoadhesive microspheres, which have the ability to reside in the gastrointestinal tract for an extended period. Cumulative percent drug release was found to be maximum for FI (91.12 %). Formulation FII found to follow Higuchi matrix with the regression value of 0.9985.39
7. Grimmettet al (2005)developed tablet containing a coated core. A tablet formulation which comprises a core containing amoxicillin trihydrate, coated with a release retarding coating, surrounded by a casing layer which includes a another active pharmaceutical substance i.e. potassium clavulanic acid. It such that there is an initial quick release of amoxicillin from the casing layer and a sustained release of amoxicillin and clavunate from the core.40
8. Ramarajuet al (2011) developed multi-layered modified release formulation comprising amoxicillin and clavulanate. The multilayered modified release formulation comprises: an immediate releae layer comprising amoxicillin and clavulanate; and a slow release layer comprising amoxicillin and one or more release retarding agents; and one or more non-release controlling inert barrier layers placed in between the immediate release layer and slow release layer and comprising one or more pharmaceutically acceptable excipients.41
9. Gregory E. Amidon, PhD. et al (2007) described different types of diluents, glidants, polymers and surfactants. The properties of excipients that ensure satisfactory and consistent performance often depend on the dosage form, the product, the manufacturing process, and the dosage form performance requirements.42
10. Igor Legenet al (2006) were worked on different pharmaceutically acceptable excipients as permeation enhancers for a low permeability drug, amoxicillin. As a model for the intestinal epithelium excised rat jejunum, mounted in side-by-side diffusion cells, was used. Among the tested pharmaceutically acceptable excipients only sodium lauryl sulfate (0.2 mg/ml) increased the permeability of amoxicillin in the mucosal-to-serosal direction, which was accompanying with the abolishment of the secretory oriented transport of amoxicillin. Other excipients (0.072 mg/ml Pluronic F68, 0.2 mg/ml Lutrol F127, 0.2 mg/ml Cremophor EL or 0.2 mg/ml Carbopol 934) have no influence on the permeability of amoxicillin.43
11. M.K.Goyalet al (2010) were preparedfloating microspheres consisting of (i) calcium silicate (CS) as porous carrier; (ii) famotidine and (iii) Hydroxypropyl methylcellulose (HPMC) and ethylcellulose (EC) as polymers. The floating microspheres were evaluated for particle size, micromeritic properties, percent drug content, in vitro floating behavior, and in vitro drug release. In Vitro Buoyancy percentage of the microspheres was found to be 98.75±3.62 At pH 1.2, drug release from floating microsphere containing amoxicillin formulation AM4 was found to be 98.87 ± 0.67 % at the end of 12 hr. While at pH 7.4, Formulation AM4 released 99.23 ± 0.94 % of drug at 12 hr respectively.44
12. Raxit Y. Mehta et al (2010) worked on the Utility of Ultra-High Viscosity Hypromellose in Extended Release Matrix Formulations. Hypromellose (HPMC) high viscosity grades K100M and K200M were used to formulate ketoprofen and metformin HCl extended release (ER) tablets. The effects of these two viscosity grades of HPMC on drug release profiles and tablet properties were investigated using 20% to 30% polymer levels. Results indicated that HPMC viscosity did not affect the drug release profiles at either polymer level. The use of HPMC K100M resulted in similar or superior tablet properties compared to HPMC K200M.45
13. KultidaSongsuranget al (2011) produced Sustained release mucoadhesive amoxicillin tablets with tolerance to acid degradation in the stomach were studied. The sustained-release tablets of amoxicillin were prepared from amoxicillin coated with ethyl cellulose (EC) and then formulated into tablets using chitosan (CS) or a mixture of CS and beta-cyclodextrin (CD) as the retard polymer. The effects of various (w/w) ratios of EC/amoxicillin, the particle sized of EC coated amoxicillin and the different (w/w) ratios of CS/CD for the retard polymer, on the amoxicillin release profile were investigated. The result showed that the release profiles of amoxicillin were greatly improved upon coating with EC, while the inclusion of CD to the CS retardant additionally prolonged the release of the drug slightly. 46
14. HN Shivakumaret al(2006) were prepared pH sensitive multi-particulate system intended to approximate the chronobiology of angina pectoris is proposed for colonic targeting. The system comprising of Eudragit S-100 coated pellets was designed for chronotherapeutic delivery of diltiazem hydrochloride. The drug loaded core pellets were produced by aqueous extrusion spheronization technique using microcrystalline cellulose as a spheronizing aid and PVP K 30 as a binder. Different coat weights of Eudragit S-100 were applied to the drug loaded pellets in an automatic coating machine to produce the pH sensitive coated pellets. In vitro dissolution studies of the coated pellets performed following pH progression method showed that the drug release from the coated pellets depended on the coat weights applied and pH of the dissolution media.47
15. MdARahmanet al(2008) prepared multiparticulate formulation of sodium paraaminosalicylate for oral administration was developed by extrusion spheronization technique. Microcrystalline cellulose was used as filler in concentration of 14.4% w/w. Pellets were coated with Eudragit L 30 D-55 using fluidized bed processor. Different weight gains of acrylic polymer were applied onto the pellets and evaluated for in vitro dissolution behavior in 0.1 N HCl for two hours and then media was changed to phosphate buffer pH 6.8. A 60% w/w coating level of Eudragit L30 D 55 has produced the most acceptable results against the gastric attack.48
16. Hagstadet al(1984) concluded that the bronchial concentration of amoxicillin (dose 500 mg) exceeded the MIC for the sensitive strains of H.influenza. the penetration of amoxicillin in to bronchial secretions was investigated by bronchioscopy.32
17. Sj: Ovallet al (1985) studied the disposition kinetics of amoxicillin and ampicillin in human volunteers. There was a tendency to more sustained plasma concentrations after amoxicillin which was also proved by a significantly lower mean plasma clearance.32
18. Watson et al (1986) studied the effect of ingesta on the systemic availability of different penicillins. In fasted dogs ampicillin showed poorer systemic avaibility than did amoxicillin, with Cmax and AUC values of less than half those of amoxicillin. The solid and liquid preparations of amoxicillin had similar bioavailability.32
19. Arancibiaet al(1987) studied the pharmacokinetics and bioavailability of a controlled release amoxicillin formulation. The results indicated that there was no correlation between the in-vitro dissolution rate and the pharmacokinetic behaviour in the body.32
20. Sugawara et al(1990) studied the transport characteristics of aminopenicillins (ampicillin and amoxicillin) and found out that the passive transport mechanism contribute a lot in the intestinal absorption of the beta lactam antibiotics.32
21. MuniyandySaravananet al(2002) studied the different batches of cephalexin extended release tablet were prepared by wet granulation method by using Eudragit L100. The effect of the concentration of Eudragit L100, microcrystalline cellulose and tablet hardness on cephalexin release was studied. The dissolution results showed that a higher amount of Eudragit in tablet composition and higher tablet hardness resulted in reduced drug release. An increased amount of microcrystalline cellulose in tablet composition resulted in enhanced drug release. Tablet composition of 13.3% w/w Eudragit L100 and 6.6 to 8% w/w microcrystalline cellulose with hardness of 7—11 kg/cm2 gave predicted release for 6 h.49
MATERIALS USED
Table: 4.1 List of ingredients used in the different formulation
|
Sr. no. |
Ingredients name |
Manufacturer |
Supplier |
|
1 |
Amoxicillin trihydrate |
DSM Antiinfective IND. |
Vaishali agencies Pvt. Ltd. |
|
2 |
HPMC K4M |
Colorcon Asia Pvt. Limited |
Colorcon Asia Pvt. Limited |
|
3 |
Ethyl cellulose (ETHOCEL STANDARD 7 PREMIUM ID48253) |
Colorcon Asia Pvt. Limited |
Colorcon Asia Pvt. Limited |
|
4 |
METHOCEL E6 PREMIUM LV HPMC |
Colorcon Asia Pvt. Limited |
Colorcon Asia Pvt. Limited |
|
5 |
PVP k-30 |
China |
Vishal pharma |
|
6 |
MCC (101) |
Gujarat microwax Ltd. |
Vishal pharma |
|
7 |
Magnesium stearate |
Komal pharmaceuticals |
Vishal pharma |
|
8 |
Talc |
Gangotrti |
Vishal pharma |
|
9 |
Coating I (OPADRY ENTERIC 940580000 WHITE)* |
Colorcon Asia Pvt. Limited |
Colorcon Asia Pvt. Limited |
|
10 |
Coating II (OPADRY ENTERIC 95K580000 WHITE)# |
Colorcon Asia Pvt. Limited |
Colorcon Asia Pvt. Limited |
|
11 |
Brilliant blue |
Asim product |
Centurion lab., baroda. |
|
12 |
Empty hard gelatin capsule shell |
Amit enterprises, pithampur. |
Gautampharmachem, Vadodara |
|
13 |
Isopropyl alcohol |
Lee changyung chemical corp. |
Shriyogeshwar trading co. |
|
14 |
Methylene chloride |
Gujarat Alkalies Chemical Ltd. |
Shriyogeshwar trading co. |
HPMC: Hydroxy Propyl Methyl Cellulose,
PVP: polyvinyl pyrrolidone,
MCC: microcrystalline cellulose
*Coating I:mixture of methacrylic acid copolymer (L-100), triethyl citrate, titanium dioxide, and talc,
#Coating II:Mixture of methacrylic acid copolymer (S-100), titanium dioxide, talc and triacetin.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
EQUIPMENTS USED:
Table 4.2: List of equipment used
|
Sr. No. |
Equipments |
Make |
|
1 |
Digital Weighing Balance |
Anameda series. Model no. model no:AA-2200 |
|
2 |
Mechanical Stirrer |
Mayura analytical pvt. Ltd. |
|
3 |
HPLC |
Agilent technologies. Model no.: 1120 compact LC |
|
4 |
Tablet Compression Machine |
Cadmach machinery co. pvt. Ltd., Type no.: CMD3-16 |
|
5 |
Tablet Hardness Tester |
Ketan, Bombay. (Monsanto) |
|
6 |
Friabilator (USP) |
Campbell electronics |
|
7 |
Dissolution Apparatus |
S.P.Automatics, Bombay. Model no:SP/DT/8 |
|
8 |
Bulk density apparatus |
Campbell electronics |
|
9 |
Digital melting point apparatus |
Veego scientific device, Model no: MP-II |
|
10 |
pH meter |
Analab instrument |
|
11 |
UV spectrometer |
Carl zeissjena, Model no.: VS U2-P. SYSTRONIC 119. |
|
12 |
Ultra sonicator |
Toshniwal process instrument pvt. Ltd. Model no.: SW4.5 |
|
13 |
Hand operated capsule filling machine |
Pam pharmaceutical and allied machinery company Pvt. Ltd. Model no.:SS072 |
ANALYTICAL METHODS
Preparation of Buffers and Reagents:
50mM of potassium phosphate monobasic buffer pH 4: Dissolve 6.8gm of potassium di-hydrogen ortho-phosphate in 1000ml water. Adjust pH 4 by using 1M of phosphoric acid.
50mM of potassium phosphate monobasic buffer pH 6: Dissolve 6.8gm of potassium di-hydrogen ortho-phosphate in 1000ml water. Adjust pH 6 by using 1M sodium hydroxide.
50mM of potassium phosphate monobasic buffer pH 6.8: Dissolve 6.8gm of potassium di-hydrogen ortho-phosphate in 1000ml water. Adjust pH 6.8 by using 1M sodium hydroxide.
1M sodium hydroxide: It prepared by dissolving 40gm of sodium hydroxide in sufficient water to produce 1000ml.
1M orthophosphoric acid: It prepared by diluting 98gm of phosphoric acid to 1000ml with water.
1) Preparation of amoxicillin trihydrate standard stock solution (1000µg/ml) in distilled water: A standard stock solution of amoxicillin trihydrate was prepared by dissolving accurately weighed 100 mg of amoxicillin trihydrate in distilled water in a 100 ml volumetric flask and the volume was made up to 100 ml by using distilled water to obtain a stock solution of 1000 mg/ml.
Determination of analytical wavelength: From the standard stock solution, 1 ml was pippetted into 10 ml volumetric flask. The volume was made up to 10 ml with distilled water. The resulting solution containing 100 mg/ml was scanned between 200 and 400 nm. The lmax was found to be 272.6nm
Calibration curve of amoxicillin trihydrate in distilled water: Accurately weighed quantity of amoxicillin trihydrate (100 mg) was dissolved in little quantity of distilled water and volume was made up to 100 ml with the same solution. Appropriate aliquots were taken into different volumetric flasks and volume was made up to 10 ml with distilled water so as to get drug concentrations of 50, 100, 150, 200, 250, 300 and 350 mg/ml. The absorbencies of these drug solutions were estimated at lmax272.6 nm. This procedure was performed in triplicate to validate the calibration curve. The data are given in the Table 4.3. A calibration curve constructed is shown in the Figure 4.1.
Table 4.3: Data for calibration curve of amoxicillin trihydrate in distilled water at 272.6 nm
|
No. |
Concentration (ppm) |
Absorbance at 272.6nm (n=3) |
|
1 |
50 |
0.133 + 0.005 |
|
2 |
100 |
0.264 + 0.003 |
|
3 |
150 |
0.395 + 0.006 |
|
4 |
200 |
0.522 + 0.000 |
|
5 |
250 |
0.652 + 0.010 |
|
6 |
300 |
0.785 + 0.007 |
|
7 |
350 |
0.914 + 0.002 |
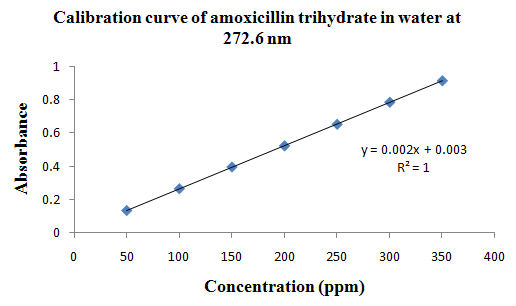
Figure 4.1: Calibration curve of amoxicillin trihydrate in distilled water.
2) Preparation of amoxicillin trihydrate standard stock solution (1000 µg/ml) in 50mM potassium phosphate monobasic buffer at 4 pH solution: A standard stock solution was prepared by dissolving accurately weighed 116 mg of amoxicillin trihydrate is equivalent to 100 mg of amoxicillin in little quantity of phosphate buffer (pH 4) solution in 100 ml volumetric flask. The volume was made up to 100 ml with phosphate buffer (pH 4) solution to obtain a stock solution of 1000 mg/ml.
Determination of analytical wavelength in phosphate buffer (pH 4) solution: From the standard stock solution, 1 ml was pippetted into 100 ml volumetric flask. The volume was made up to 100 ml with phosphate buffer solution, pH 4. The resulting solution containing 10 mg/ml was scanned between 200 and 400 nm. The lmaxwas found to be 272.2.
Calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 4: From the stock solution, appropriate aliquots were taken from the above solution into different volumetric flasks and made up to 10 ml with phosphate buffer solution pH 4, so as to get drug concentrations ranging from 50 to 350 mg/ml. The absorbencies of these drug solutions were estimated at lmax272.2 nm. This procedure was performed in triplicate to validate the calibration curve. The data are given in the Table 4.4. A calibration curve constructed is shown in the Figure 4.2.
Table 4.4: Data for calibration curve of amoxicillin trihydrate in phosphate buffer, pH 4 at 272.2 nm
|
No. |
Concentration (ppm) |
Absorbance at 272.2nm (n=3) |
|
1 |
50 |
0.161 + 0.010 |
|
2 |
100 |
0.319 + 0.002 |
|
3 |
150 |
0.470 + 0.007 |
|
4 |
200 |
0.622 + 0.003 |
|
5 |
250 |
0.776 + 0.001 |
|
6 |
300 |
0.929 + 0.006 |
|
7 |
350 |
1.080 + 0.002 |
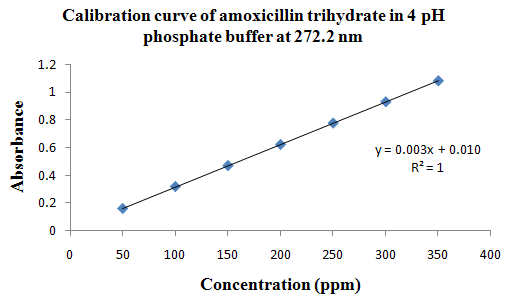
Figure 4.2: Calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 4.
3) Preparation of amoxicillin trihydrate standard stock solution (1000 µg/ml) in 50mM potassium phosphate monobasic buffer at 6 pH solutions: A standard stock solution was prepared by dissolving accurately weighed 116 mg of amoxicillin trihydrate equivalent to 100 mg of amoxicillin in little quantity of phosphate buffer (pH 6) solution in 100 ml volumetric flask. The volume was made up to 100 ml with phosphate buffer (pH 6) solution to obtain a stock solution of 1000 mg/ml.
Determination of analytical wavelength in phosphate buffer (pH 6) solution: From the standard stock solution, 1 ml was pippetted into 100 ml volumetric flask. The volume was made up to 100 ml with phosphate buffer solution, pH 6. The resulting solution containing 10 mg/ml was scanned between 200 and 400 nm. The lmaxwas found to be 272.8.
Calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 6: From the stock solution, appropriate aliquots were taken from the above solution into different volumetric flasks and made up to 10 ml with phosphate buffer solution pH 6, so as to get drug concentrations ranging from 50 to 350 mg/ml. The absorbencies of these drug solutions were estimated at lmax272.8 nm. This procedure was performed in triplicate to validate the calibration curve. The data are given in the Table 4.5. A calibration curve constructed is shown in the Figure 4.3.
Table 4.5: Data for calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 6 at 272.8 nm
|
No. |
Concentration (ppm) |
Absorbance at 272.2nm (n=3) |
|
1 |
50 |
0.164 + 0.006 |
|
2 |
100 |
0.306 + 0.001 |
|
3 |
150 |
0.470 + 0.003 |
|
4 |
200 |
0.625 + 0.008 |
|
5 |
250 |
0.773 + 0.010 |
|
6 |
300 |
0.924 + 0.007 |
|
7 |
350 |
1.078 + 0.004 |
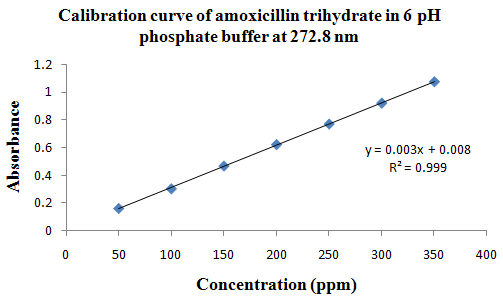
Figure 4.3: Calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 6.
4) Preparation of amoxicillin trihydrate standard stock solution (1000 µg/ml) in 50mM potassium phosphate monobasic buffer at 6.8 pH solution: A standard stock solution was prepared by dissolving accurately weighed 116 mg of amoxicillin trihydrate equivalent to 100 mg of amoxicillin in little quantity of phosphate buffer (pH 6.8) solution in 100 ml volumetric flask. The volume was made up to 100 ml with phosphate buffer (pH 6.8) solution to obtain a stock solution of 1000 mg/ml.
Determination of analytical wavelength in phosphate buffer (pH 6.8) solution: From the standard stock solution, 1 ml was pippetted into 100 ml volumetric flask. The volume was made up to 100 ml with phosphate buffer solution, pH 6.8. The resulting solution containing 10 mg/ml was scanned between 200 and 400 nm. The lmaxwas found to be 272.6.
Calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 6.8: From the stock solution, appropriate aliquots were taken from the above solution into different volumetric flasks and made up to 10 ml with phosphate buffer solution pH 6.8, so as to get drug concentrations ranging from 50 to 350 mg/ml. The absorbencies of these drug solutions were estimated at lmax272.6 nm. This procedure was performed in triplicate to validate the calibration curve. The data are given in the Table 4.6. A calibration curve constructed is shown in the Figure 4.4.
Table 4.6: Data for calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 6.8 at 272.6 nm
|
No. |
Concentration (ppm) |
Absorbance at 272.6nm (n=3) |
|
1 |
50 |
0.167 + 0.005 |
|
2 |
100 |
0.327 + 0.000 |
|
3 |
150 |
0.493 + 0.003 |
|
4 |
200 |
0.661 + 0.007 |
|
5 |
250 |
0.807 + 0.001 |
|
6 |
300 |
0.977 + 0.000 |
|
7 |
350 |
1.133 + 0.002 |
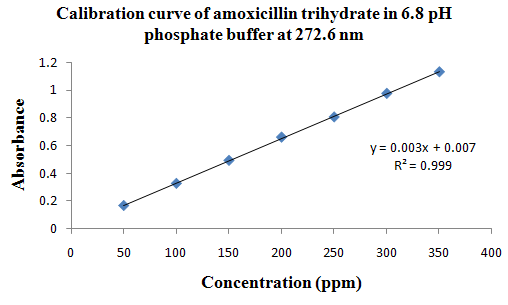
Figure 4.4: Calibration curve of amoxicillin trihydrate in phosphate buffer solution, pH 6.8.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
PREFORMULATION STUDIES 6, 50
It can be defined as an investigation of physical and chemical properties of a drug substance alone and when combined with excipients. The overall objective of preformulation testing is to generate information useful to the formulation in developing stable and bioavailable dosage forms that can be mass-produced. The type of information needed will depend on the dosage form to be developed.
Preformulation studies are the first step in the rational development of dosage form of a drug substance. The objective of preformulation studies is to develop a portfolio of information about the drug substance, so that this information will be useful to develop different dosage forms. Preformulation can be defined as investigation of physical and chemical properties of drug substance with excipients.
Preformulation investigations are designed to identify those physicochemical properties and excipients that may influence the formulation design, method of manufacture, and pharmacokinetic-biopharmaceutical properties of the resulting product.
A. API characterization
a) Organoleptic characteristics
b) Determination of melting point
c) Angle of Repose (funnel method)
d) Bulk Density
e) Compressibility Index (Carr’s Index)
f) Hausner Ratio
B. Excipients characterization
C. Drug-excipients Compatibility studies
A. API characterization:
API characterization like, determination of melting point range, physico mechanical characterization like Bulk density, True density, Haussner’s Ratio, Carr’s/Compressibility Index etc.
a) Organoleptic characteristics:
The color, odor, and taste of the drug were characterized and recorded using descriptive terminology.
b) Determination of melting point: Melting point was determined by taking small amount of amoxicillin trihydrate in a capillary tube closed at one end. The capillary tube was placed in an electrically operated melting point apparatus and the temperature at which the drug melts was recorded. This was performed thrice and average value was noted.
c) Angle of Repose: (funnel method)
The angle of repose values of amoxicillin trihydrate determined by the funnel method. The accurately weighed drug was taken in a funnel. The height of the funnel was adjusted in such a way that the tip of the funnel just touches the apex of the heap of the powder. The powder was allowed to flow through the funnel freely onto the surface. The diameter of the powder cone was measured and angle of repose was calculated using the following equation
ø = tan-1 (h/r) ----------- (6)
where, h = the height of the powder cone ;
r = the radius of the powder cone
Flow properties for different values of angle of repose are given below.
Table 4.7: Comparison between Angle of Repose and flow property
PREFORMULATION STUDIES 6, 50
|
Angle of Repose (degrees) |
Flow property |
|
25-30 |
Excellent |
|
31-35 |
Good |
|
36-40 |
Fair- aid not needed |
|
41-45 |
Passable-may hang up |
|
46-55 |
Poor-must agitate, vibrate |
|
56-65 |
Very poor |
|
Very, very poor |
>66 |
d) Bulk Density:
Loose bulk density (LBD) and tapped bulk density (TBD) of amoxicillin trihydrate determined using bulk density apparatus.Accurately weighed drug was placed in a 100 ml graduated measuring cylinder. Initial volume was observed. The cylinder was tapped initially 500 times from a distance of 14 + 2 mm. The tapped volume (Va) was measured to the nearest graduated unit. The tapping was repeated additional 750 times. Again the tapped volume was measured to the nearest graduated unit. The same thing was done for powder blends of the tablets. The LBD and TBD were calculated in g per ml using following formula;
LBD = weight of the powder / volume of the packing ------------- (7)
TBD = weight of the powder / tapped volume of the packing ----- (8)
e) Compressibility Index (Carr’s Index)
The compressibility of the powder was determined by the Carr’s compressibility index.
Carr’s index (%) = [(TBD – LBD) x 100] / TBD ----------- (9)
Table 4.8: Comparison between % Compressibility and flow property
|
% Compressibility |
Flow ability |
|
<10 |
Excellent |
|
11-15 |
Good |
|
16-20 |
Fair |
|
21-25 |
Passable |
|
26-31 |
Poor |
|
32-37 |
Very poor |
|
>38 |
Very, very poor |
f) Hausner Ratio
The Hausner ratio of the powder was determined by the following equation.
Hausner ratio = TBD / LBD ----------- (10)
Table 4.9: Comparison between Hausner Ratio value and flow property
|
Hausner Ratio |
Flow character |
|
1.00-1.11 |
Excellent |
|
1.12-1.18 |
Good |
|
1.19-1.25 |
Fair |
|
1.26-1.34 |
Passable |
|
1.35-1.45 |
Poor |
|
1.46-1.59 |
Very poor |
|
>1.60 |
Very, very poor |
B. Excipients characterization
Excipients characterization like bulk density, tapped density, compressibility index and hausner ratio evaluated for its flow property and compressibility.
C. Drug-excipients Compatibility studies
In the preparation, drug and polymer may interact as they are in close contact with each other, which could lead to the instability of drug. Preformulation studies regarding the drug-polymer interaction are therefore very critical in selecting appropriate polymers. FT-IR spectroscopy was employed to ascertain the compatibility between amoxicillin trihydrate and the selected polymers. The pure drug and drug with excipient were scanned separately.
Procedure: Potassium bromide was mixed with drug and/or polymer in 9:1 ratio and the spectra were taken. FT-IR spectrum of amoxicillin trihydrate was compared with FT-IR spectra of amoxicillin trihydrate with polymer. Disappearance of amoxicillin trihydrate peaks or shifting of peak in any of the spectra was studied.
FORMULATION DEVELOPMENT
The theoretical drug release profile calculation is important to evaluate the formulation with respect to release rates and to ascertain whether it releases the drug in a predetermined manner.Theoretically drug release profile was calculated by using the pharmacokinetic data.
Development of the formulation in the present study was mainly based on the type and concentration of polymers and the properties of the drug. Various polymers in different combinations were used so as to get dissolution profile that matches with the theoretical drug release profile. Furthermore it compared to different hardness to achieve desired drug release profile.
Manufacture of amoxicillin trihydratedosage forms:
Amoxicillin trihydrate tablets and capsules (775 mg) were manufactured for the thirteen batches T1 to T13 and C1 to C6 using the ingredients mentioned in the Table 4.10 and Table 4.11 respectively.
Table 4.10: Formulations of amoxicillin trihydrate tablet
|
INGREDIENTS |
FORMULATIONS |
||||||||||||
|
T1 |
T2 |
T3 |
T4 |
T5 |
T6 |
T7 |
T8 |
T9 |
T10 |
T11 |
T12 |
T13 |
|
|
Amoxicillin trihydrate(mg) |
775 |
775 |
775 |
775 |
775 |
775 |
775 |
775 |
775 |
775 |
775 |
775 |
775 |
|
Ethyl cellulose (%) |
- |
- |
- |
8 |
6 |
4 |
2 |
95:5 |
85:15 |
75:25 |
65:35 |
55:45 |
50:50 |
|
METHOCEL (HPMC) |
- |
- |
- |
- |
- |
- |
- |
||||||
|
PVP K-30 (mg) |
23.16 |
- |
- |
- |
- |
- |
- |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
1.5 |
|
MCC (mg) |
26.8 |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
|
HPMC K4M (mg) |
- |
291 |
291 |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
|
Coating I (%)* |
8 |
- |
8 |
8 |
8 |
8 |
8 |
- |
- |
- |
- |
- |
- |
|
Coating II (%)# |
8 |
- |
8 |
8 |
8 |
8 |
8 |
- |
- |
- |
- |
- |
- |
|
Brilliant blue |
qs |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
|
Magnesium stearate (mg) |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
|
Total (gm) |
1.065 |
1.212 |
1.300 |
1.044 |
1.025 |
1.006 |
0.988 |
0.99 |
0.99 |
0.99 |
0.99 |
0.99 |
0.99 |
HPMC: Hydroxy Propyl Methyl Cellulose,
PVP: polyvinyl pyrrolidone,
MCC: microcrystalline cellulose
*Coating I:mixture of methacrylic acid copolymer (L-100), triethyl citrate, titanium dioxide, and talc.
#Coating II:mixture of methacrylic acid copolymer (S-100), titanium dioxide, talc and triacetin
Table 4.11: Formulations of amoxicillin trihydrate capsule
|
INGREDIENTS |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
|
Amoxicillin trihydrate(mg) |
775 |
775 |
775 |
775 |
775 |
775 |
|
Ethyl cellulose (%) |
- |
- |
- |
8 |
15 |
20 |
|
PVP K-30 (mg) |
23.16 |
- |
- |
- |
1.5 |
1.5 |
|
MCC (mg) |
26.8 |
- |
- |
- |
- |
- |
|
HPMC K4M (mg) |
- |
291 |
291 |
- |
- |
- |
|
Coating I (%)* |
8 |
- |
8 |
8 |
- |
- |
|
Coating II (%)# |
8 |
- |
8 |
8 |
- |
- |
|
Brilliant blue |
Qs |
- |
- |
- |
- |
- |
|
Magnesium stearate (mg) |
5 |
5 |
5 |
5 |
5 |
5 |
|
Total (gm) |
1.065 |
1.212 |
1.300 |
1.044 |
1.054 |
1.10 |
HPMC: Hydroxy Propyl Methyl Cellulose,
PVP: polyvinyl pyrrolidone,
MCC: microcrystalline cellulose
*Coating I: mixture of methacrylic acid copolymer (L-100), triethyl citrate, titanium dioxide, and talc.
#Coating II:mixture of methacrylic acid copolymer (S-100), titanium dioxide, talc andtriacetin
Table 4.12: List of materials are used in present study with use
|
Sr. no. |
Ingredients name |
Function |
|
1 |
Amoxicillin trihydrate |
Drug |
|
2 |
HPMC K4M |
Delayed release Polymer |
|
3 |
Ethyl cellulose (ETHOCEL STANDARD 7 PREMIUM ID48253) |
Delayed release Polymer |
|
4 |
METHOCEL E6 PREMIUM LV HPMC |
Modified release polymer |
|
5 |
PVP k-30 |
Binder |
|
6 |
MCC (101) |
Diluent |
|
7 |
Magnesium stearate |
Lubricant |
|
8 |
Talc |
Lubricant |
|
9 |
Coating I (OPADRY ENTERIC 940580000 WHITE) |
Delayed release coating |
|
10 |
Coating II (OPADRY ENTERIC 95K580000 WHITE) |
Delayed release coating |
|
11 |
Brilliant blue |
Colouring agent |
|
12 |
Isopropyl alcohol |
Solvent |
|
13 |
Methylene chloride |
Solvent |
Amoxicillin trihydrate is active pharmaceutical ingredient. Pvp k30 act as binder in all formulations. Ethyl cellulose, HPMC K4M, methocel is polymers used to modified the drug release from dosage form. Coating I and coating II are the coating material which modified the release of drug from dosage form. Magnesium stearate used as a lubricant. Brilliant blue used as a colouring agent in the formulation.
Procedure: T121
In the formulation procedure of for the batch T1, total dose of amoxicillin trihydrate divided in three parts. Amoxicillin trihydrate divided as 45%, 30% and 25% in part I, part II and part III respectively.
Part I (immediate release granules) containing amoxicillin trihydrate and MCC and it binds with the PVP K30 which is dissolved in the mixture of isopropyl alcohol and methylene dichloride.Coarse screening of wet mass using a suitable sieve (10# screens).Drying of moist granules under sunlight for 15minutes. Screening of dry granules through a suitable sieve (20 # screen). From part I granules drug release in the stomach.
Part II (1st delayed release granules) granules containing amoxicillin trihydrate and MCC, it binds with the PVP K30 which is dissolved in the mixture of isopropyl alcohol and methylene dichloride. These prepared granules coat with the extended release polymer coating I. From part II granules drug release in the upper small intestine.
Part III (2nd delayed release granules) granules containing amoxicillin trihydrate and MCC, it binds with the PVP K30 which is dissolved in the mixture of isopropyl alcohol and methylene dichloride. These prepared granules coat with the extended release polymer coating II. From part II granules drug release in the upper small intestine.
It prepared by the taking all different immediate release granules, 1st delay release granules and 2nd delay release granules and mix with lubricant. Compress it in a tablet form by using D-tooling 16- station compression machine.
Procedure T2:
Dry mixing of drug and polymer like HPMC for 10 min.Lubricate it by Blend of tablet mix with lubricant for 10min.Compress the blend into tablets using D-Tooling 16- station compression machine.
Procedure T3 to T7:
Dry mixing of drug and polymer for 10 min. granules prepared by wet granulation method. Mixture of drug and polymer binds with Isopropyl alcohol.Coarse screening of wet mass using a suitable sieve (10 # screens).Dry the granules under sunlight for 10min. Screening of dry granules by passes it from sieve (20 # screens).Divide the granules ratio as per trial basis release of drug required. All granules divide in to three portions. First portion is the immediate release of drug and other two for the delayed release of drug. Coating the two portion of delayed release granules with the different coating material. All granules were mixed and lubricate with magnesium stearate. Compress the blend into tablets using D-Tooling 16- station compression machine.
Procedure T8 to T13:
Dry mixing of drug and polymer for 10 min. granules prepared by wet granulation method. Mixture of drug and polymer binds with Isopropyl alcohol.Coarse screening of wet mass using a suitable sieve (10 # screens).Dry the granules under sunlight for 10min. Screening of dry granules by passes it from sieve (20 # screens).Lubricate with magnesium stearate. Compress the blend into tablets using D-Tooling 16- station compression machine.
Procedure C1 to C4 (capsules):
Granules preparations of the capsule from batches C1 to C4 are same as the tablet. And those granules fill in to two 00size empty hard gelatin capsule shell by using hand operated capsule filling machine.
Procedure C5 and C6 (capsules):
Dry mixing of drug and polymer, given in table 4.11 for 10 min. granules prepared by wet granulation method. Mixture of drug and polymer binds with Isopropyl alcohol.Coarse screening of wet mass using a suitable sieve (10 # screens).Dry the granules under sunlight for 10min. Screening of dry granules by passes it from sieve (20 # screens).Lubricate with magnesium stearate. Compress the blend into tablets with higher hardness using D-Tooling 16- station compression machine.Crush tablet and pass it from sieve 14#. Fill it in a two 00size hard gelatin capsule shell by using hand operated capsule filling machine.
EVALUATION OF GRANULES
Granules flow properties determined like angle of repose, Bulk density, Tapped density, Haussner’s Ratio, Carr’s/Compressibility Index. All these measured as per above methods.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
EVALUATION OF AMOXICILLIN TRIHYDRATE TABLETS51
The tablets after punching of every batch were evaluated for in process and finished product quality control tests i.e. appearance, dimensions (diameter and thickness), weight variation, hardness, friability, drug content, and in vitro drug release.
1) Appearance:
The tablets were checked for presence of cracks, depressions, pinholes etc if any, uniformity of the colour, and the polish of the tablet.
2) Thickness:
The thickness of the tablet from production run to production is carefully controlled. Thickness can vary with no change in weight due to difference in the dosing of the granulation and the pressure applied to the tablets as well as the speed of the tablet compression. Tablet thickness also becomes an important characteristics packaging. If thickness varies throughout the lot, the result will vary in the count.
3) Hardness:
The resistance to the tablet to chipping, abrasion or breakage under condition of storage, transportation and handling before usage depends upon its hardness. i.e. it measure the force required to break the tablet when the force is applied diametrically to the tablet. The force is measured in kilograms when used in production, hardness of 4 kg is considered to be minimum for a satisfactory tablet. Hardness determinations are made throughout the tablet runs to determine the need for pressure adjustments on the tablet compression machine.
4) Weight variation:
The volumetric fill of the die cavity determines the weight of the compressed tablet. In setting up the tablet machine the fill adjusted to give the desired tablet weight. After the tablet machine is in operation the weights of the tablets were checked routinely to ensure that proper weight of tablet are being made.
To study weight variation, 20 tablets of each formulation were weighed using an electronic balance and the test was performed as follow: Twenty tablets were weighed individually and all together. Average weight was calculated from the total weight of all tablets. The individual weights were compared with the average weight. The percentage difference in the weight variation should be within the permissible limits (±5%). The percent deviation was calculated using the formula:
(Individual weight – Average weight)
Percentage Deviation = ---------------------------------- x 100
Average weight
-- (11)
5) Friability:
Friability of tablets was determined using Roche friabilator. This device subject the tablets to the combined effect of abrasions and shock in a plastic chamber revolving at 25 rpm and dropping the tablets at a height of six inches in each revolution. Preweighed sample of tablets was placed in the friabilator and were subjected to 100 revolutions. Tablets were dedusted using a soft muslin cloth and reweighed.
This in process quality control test is performed to ensure the ability of tablets to withstand the shocks during processing, handling, transportation, and shipment. Permitted friability limit is 1.0 %. The friability is given by the formula:
(Winitial-Wfinal)
Friability = ------------------------ x 100 ----- (12)
Winitial
6) Assay: (as per IP)
HPLC parameters:
Column: C18, ODS, 250nm x 4.6nm, 5μ
Mobile Phase: solvent mixture: Acetonitrile (96:4)
Solvent mixture: 6.8gm monobasic potassium phosphate in 1000 ml water. Add potassium hy droxide and adjust the pH 4.5.
Flow rate: 1.5 ml/minInjection Volume: 20 μL
Detection: At 230nm
Standard preparation:
Weigh and transfer accurately about 290mg amoxicillin trihydrate in 100ml solvent mixture, and shaking with the aid of ultrasound to dissolve. Pipette out 2ml into 25ml volumetric flask and Make up the volume with solvent mixture and mix.
Sample Preparation:
Weigh and powder 5 tablet. Weigh accurately a quantity of the powdered tablet containing 290mg amoxicillin trihydrate in 100ml solvent mixture, and shaking with the aid of ultrasound to dissolve. Pipette out 2ml into 25ml volumetric flask and Make up the volume with solvent mixture and mix.
Procedure:
Inject blank (mobile phase) and record the chromatogram.
Inject the standard preparation (For RT comparison) and record the chromatogram.
Inject a sample preparation and record the chromatogram.
Formula:
Gm/tablet = Standard area * standard weight * potency * average wt of tab.
Sample area sample weight 100 ------------- (13)
% = gm/tablet * 100 ------------ (14)
Theoretical dose
7) In-vitrodrug release:52 (USFDA)
In vitrodrug release of the samples was carried out using USP – type II dissolution apparatus (paddle type). The dissolution medium, 900ml of 50mM of potassium phosphate monobasic buffer at pH 4 for 0-2hrs, at pH 6 for 2-4hrs and at pH 6.8 for 4 to 24hrs or 900ml water was placed into the dissolution flask maintaining the temperature of 37 + 0.5 oC and rpm of 100. One amoxicillin trihydrate tablet was placed in each flask of dissolution apparatus. The apparatus was allowed to run for 24 hours. Samples measuring 10 ml were withdrawn after different time interval. Samples were filtered through whatman filter paper. The fresh dissolution medium was replaced every time with the same quantity of the sample. The collected samples were analyzed on UV spectrometer using dissolution medium as blank. The cumulative percentage drug release was calculated.
Dissolution Parameter:
Medium: 900mL; 50mM potassium phosphate monobasic buffer at pH 4, 6, 6.8 or 900 ml water.
Apparatus: USP-II (Paddle)
RPM:100
Temperature: 37°C ± 0.5°C
Comparison of Dissolution Profiles53,54
The dissolution profile comparison may be carried out using model independent or model dependent method. A simple model independent approach uses a difference factor (f1) and a similarity factor (f2) to compare dissolution profiles.
St=1n(Rt-Tt)
f1= -------------------- x 100 -----------(15)
St=1nRt
f2= 50 x log {[1+(1/n) St=1n(Rt – Tt)2]-0.5 x 100} ------- (16)
Where, Rtand Tt represent the average percent dissolved at time t for reference and test, respectively, and n is the number of time points tested. Dissolution profile was considered satisfactory if f1 values lies below 15 (nearing zero) and f2 value lies more than 50 (nearing 100).
The model independent method is most suitable for dissolution profile comparison when three to four or more dissolution time points are available. Comparison of the dissolution profiles of the prepared tablets with theoretical drug release profile was carried out.
Reproducibility
To verify the process for its reproducibility, the tablets of the optimized batch were manufactured. The tablets were evaluated for dissolution as the rest of the parameters were proved to be reproducible and compared with tablets of the earlier batch.
Influence of hardness on drug release49, 55
One could relate variation in compression force to a change in the porosity of the tablet. Compression force is expected to have little influence on drug release rate. Once sufficient tablet hardness suitable for processing and handling is achieved, tablet hardness would have little further effect on drug release profile.
Mathematical-model of amoxicillin trihydrate tablet56
The results obtaining in vitro release studies were plotted in different models of data treatment as follows: Two commonly used deconvolution methods for estimating the apparent in vitro drug absorption profiles following oral administration of a dosage form are
1) Dependent-model method
2) In dependent-model method (data analysis)
1) Dependent-model method:
a) Zero Order Kinetic
It describes the system in which the drug release rate is independent of its concentration.
Qt = Qo + Ko t ----------- (17)
Where,
Qt= Amount of drug dissolved in time t
Qo = Initial amount of drug in the solution, which is often zero and
Ko= zero order release constant.
If the zero order drug release kinetic is obeyed, then a plot of Qt versus t will give a straight line with a slope of Ko and an intercept at zero.
b) First Order Kinetic
It describes the drug release from the systems in which the release rate is concentration dependent.
logQt = log Qo + kt/ 2.303 ----------- (18)
Where, Qt = amount of drug released in time t.
Qo = initial amount of drug in the solution
k = first order release constant
If the first order drug release kinetic is obeyed, then a plot of log (Qo- Qt) versus t will be straight line with a slope of kt/ 2.303 and an intercept at t=0 of log Qo
c) Higuchi Model
It describes the fraction of drug release from a matrix is proportional to square root of time.
Mt / M∞= kHt1/2 ---------- (19)
Where,
Mt and M∞ = cumulative amounts of drug release at time t and infinite time,
kH = Higuchi dissolution constant reflection formulation characteristics.
If the Higuchi model of drug release (i.e. Fickian diffusion) is obeyed, then a plot of Mt / M∞ versus t1/2 will be straight line with slope of kH.
d) Hixson and Crowell cube root law
Hixson and Crowell recognized that theparticlesi regular area is proportional to the cube root of its volume. They derived the equation:
W01/3- Wt1/3 = κ t ---------- (20)
Where, W0= initial amount of drug in the pharmaceutical dosage form,
Wt = remaining amountof drug in the pharmaceutical dosage form at time t
κ (kappa) = constant incorporating the surface-volume relation.
The equation describes therelease from systems where there is a change in surface area and diameter of particles or tablets. To study the release kinetics, data obtained from in vitrodrug release studies were plotted as cube root of drug percentage remaining in matrix versus time. Application: This expression applies to pharmaceutical dosage form such as tablets, where the dissolution occurs in planes that are parallel to the drug surface if the tablet dimensions diminish proportionally, in such a manner that the initial geometrical form keeps constant all the time.
e) Korsmeyer-Peppas model (Power Law)
The power law describes the drug release from the polymeric system in which release deviates from Fickian diffusion, as expressed in following equation.
Mt / M∞= ktn --------- (21)
log [Mt / M∞] = log k + n log t ---------- (22)
Where,
Mt and M∞ = cumulative amounts of drug release at time t and infinite time (i.e. fraction of drug release at time t),
k = constant incorporating structural and geometrical characteristics of CR device,
n = diffusional release exponent indicative of the mechanism of drug release for drug dissolution.
To characterize the release mechanism, the dissolution data {Mt / M∞ < 0.6} are evaluated. A plot of log {Mt / M∞} versus log t will be linear with slope of n and intercept gives the value of log k. Antilog of log k gives the value of k.
Peepas used the n value in order to characterize different release mechanisms as shown in the table below,
Table 4.13 Different Release Mechanisms
|
Slope ( n ) |
Mechanism |
|
<0.5 |
Fickian diffusion (Higuchi Matrix) |
|
0.5 <n< 1 |
Non-Fickian diffusion |
|
1 |
Case II transport |
Stability Studies57,58
Stability is defined as the ability of a particular drug or dosage form in a specific container to remain within its physical, chemical, therapeutic, and toxicological specifications. Drug decomposition or degradation occurs during stability, because of chemical alteration of the active ingredients or due to product instability, lowering the concentration of the drug in the dosage form. The stability of pharmaceutical preparation should be evaluated by accelerated stability studies. The optimized formulation of amoxicillin trihydrate tablets was selected for the stability studies. The accelerated stability studies were carried out according to ICH guidelines by storing the samples at 40 + 2 oC and 75 + 5% RH for 1 month.The tablets were evaluated for hardness, drug content, and dissolution study and compared with tablets which were evaluated immediately after manufacturing. Table 4.13 shows different temperatures and period of stability testing.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table 4.14 ICH guidelines for stability study
|
Study |
Storage condition |
Minimum time period covered |
|
Long term |
25°C±2°C / 60%±5% RH |
12 months |
|
Intermediate |
30°C±2°C / 65%±5% RH |
6 months |
|
Accelerated |
40°C±2°C / 75%±5% RH |
3 months |
EVALUATION OF AMOXICILLIN TRIHYDRATE CAPSULES51
The capsules after filling of every batch were evaluated for in process and finished product quality control tests i.e. appearance, weight variation, assay, and in vitro drug release.
Table 4.15: Test, Specification and Result of empty hard gelatin capsule shell
|
Test |
Specification |
Method |
Result |
|
|
PHYSICAL TESTS |
||||
|
Description |
Cylindrical, transperant body and brilliant blue cap, telescopic capsules |
I.P. |
Complies |
|
|
Average weight of capsule |
119mg + 10% |
I.P. |
120.0 mg |
|
|
Disintegration time |
Less than 15minutes |
I.P. |
8.05min. |
|
|
Loss on drying |
12.5% - 16.0% |
I.P/USP |
14-15% |
|
|
Self locking feature |
Shell and body lock together and are not readily separated |
EP |
Complies |
|
|
Brittleness |
Shell deforms under pressure but dies not crack |
EP |
Complies |
|
|
DIMENTIONS |
||||
|
|
Cap |
body |
|
|
|
Length |
11.8 + 0.4mm |
20.3 + 0.4mm |
I.P |
Complies |
|
Double wall thickness |
0.210 + 0.4mm |
0.206 + 0.014mm |
I.P |
Complies |
|
Out side diameter |
8.53 + 0.03mm |
8.21 + 0.03mm |
I.P |
Complies |
|
Closed joined length |
23.5 + 0.04mm |
EP |
Complies |
|
1) weight variation: (as per IP)
Weigh an intact capsule. Open it without losing any part of the shell and remove the contents as completely as possible. Weigh the shell. The difference between the weighings gives the weight of the contents. Repeat the procedure with another 19 capsules. Percentage deviation should be + 7.5%.
Net weight = Filled capsule weight – Empty capsule weight
(Net weight – Average weight)
Percentage Deviation = --------------------------------------------- x 100 ------ (23)
Average weight
2) Assay: (as per IP)
HPLC parameters:
Column: C18, ODS, 250nm x 4.6nm, 5μ
Mobile Phase: solvent mixture : Acetonitrile (96:4)
Solvent mixture: 6.8gm monobasic potassium phosphate in 1000 ml water. Add potassium hydroxide and adjust the pH 4.5.
Flow rate: 1.5 ml/min
Injection Volume: 20 μL
Detection: At 230nm
Standard preparation:
Weigh and transfer accurately about 290mg amoxicillin trihydrate in 100ml solvent mixture, and shaking with the aid of ultrasound to dissolve. Pipette out 2ml into 25ml volumetric flask and Make up the volume with solvent mixture and mix.
Sample Preparation:
Weigh accurately a quantity of the powdered granules from 5 capsules containing 290mg amoxicillin trihydrate in 100ml solvent mixture, and shaking with the aid of ultrasound to dissolve. Pipette out 2ml into 25ml volumetric flask and Make up the volume with solvent mixture and mix.
Procedure:
Inject blank (mobile phase) and record the chromatogram.
Inject the standard preparation (For RT comparison) and record the chromatogram.
Inject a sample preparation and record the chromatogram.
Formula:
Gm/capsule = Std area* std weight *potency * average net wt of capsule.
Sample area samplewt100 -------- (24)
% = gm/capsule * 100 --------- (25)
Theoretical dose
3) In-vitrodrug release
In vitrodrug release of the samples was carried out using USP – type II dissolution apparatus (paddle type). The dissolution medium, 900ml of 50mM of potassium phosphate monobasic buffer at pH 4 for 0-2hrs, at pH 6 for 2-4hrs and at pH 6.8 for 4 to 24hrs or 900ml water was placed into the dissolution flask maintaining the temperature of 37 + 0.5 oC and rpm of 100. One amoxicillin trihydrate capsule was placed in each flask of dissolution apparatus. The apparatus was allowed to run for 24 hours. Samples measuring 10 ml were withdrawn after different time interval. Samples were filtered through whatman filter paper. The fresh dissolution medium was replaced every time with the same quantity of the sample. The collected samples were analyzed on UV spectrometer using dissolution medium as blank. The cumulative percentage drug release was calculated.
Dissolution Parameter:
Medium: 900mL; 50mM potassium phosphate monobasic buffer at pH 4, 6, 6.8 or 900 ml water
Apparatus: USP-II (Paddle)
RPM:100
Temperature: 37°C ± 0.5°C
The results obtained and the discussion there upon was described in the next chapter “Results and Discussion”.
Dosage forms of amoxicillin trihydrate were developed with a view to deliver the drugs up to 24hrs. Analytical method was developed, preformulation studies were conducted, formulations were developed, and stability studies were conducted. MS-Excel and MS Word were used for calculations including graphs and word processing, respectively. The details of results and discussion are given in the following sections.
PREFORMULATION STUDIES
The following preformulation studies were performed on Amoxicillin Trihydrate and excipients.
A. API characterization:
Organoleptic characteristics:
The color, odor, and taste of the drug were characterized and recorded using descriptive terminology. The resultswere shown in the Table no. 18.
Table 5.1: Results of Organoleptic properties
Properties |
Results |
|
Description |
Crystalline powder |
|
Taste |
Slightly bitter |
|
Odor |
Odorless |
|
Color |
White color |
Melting point determination:
Melting point of amoxicillin trihydrate was found to be 193.5± 0 .3 (n = 3). This value is same as that of the literature citation 194oC.
Physical Properties of Amoxicillin trihydrate:
Interparticulate interactions influence the bulking properties of powder. A comparison of the bulk density and tapped density can give a measure of the relative importance of this interaction in a given powder; such a comparison is often used as an index of the ability of the powder to flow. The bulk density and tapped density were found to be 0.6058 and0.833 gm/ml, respectively.
Flow property is important for the flowing from the hopper of the compression machine during formulation. Angle of repose is one of parameter to determine flow property of powder. Angle of repose of amoxicillin was found to be 42.610.
A simple indication of ease with which a material can be induced to flow is given by application of a compressibility index. The value for compressibility index of amoxicillin trihydrate was found to be 27.27% and Hauser’s ratio of 1.37. Therefore, amoxicillin trihydrate have poor flow property. Physical properties of amoxicillin trihydrate; like bulk density, tapped density, %compressibility, and Hausner ratio results are shown in the Table 5.2.
Table 5.2: characterization of amoxicillin trihydrate as Preformulation study
|
Characterization |
Observation |
|
a) Angle of repose |
42.610 |
|
b) Bulk density (gm/ml) |
0.6058 |
|
c)Tapped density (gm/ml) |
0.833 |
|
d) Compressibility Index (%) |
27.27 |
|
e) Hausner ratio |
1.37 |
|
Flow property |
Poor |
B. Excipients characterization
Excipients characterization like bulk density, tapped density, compressibility index and hausner ratio evaluated for its flow property and compressibility.
Table 5.3:Comparative B.D, T.D, % CI, HR for Excipients
|
Sr No. |
Ingredient |
B.D. (g/ml) |
T.D. (g/ml) |
%C.I. |
H.R. |
Inference |
|
1 |
HPMC K4M |
0.341 |
0.557 |
38.779 |
1.63343 |
Very, very Poor |
|
2 |
METHOCEL |
0.345 |
0.562 |
38.612 |
1.628 |
Very, very Poor |
|
3 |
EC |
0.238 |
0.298 |
20.00 |
1.250 |
Fair |
|
4 |
PVP k-30 |
0.34 |
0.46 |
26.087 |
1.35294 |
Poor |
|
5 |
MCC 101 |
0.32 |
0.45 |
28.888 |
1.40625 |
Poor |
|
6 |
Magnesium Stearate |
0.231 |
0.298 |
22.497 |
1.29028 |
Fair |
|
7 |
Coating I* |
0.35 |
0.42 |
16.666 |
1.200 |
Fair |
|
8 |
Coating II# |
0.30 |
0.369 |
18.94 |
1.233 |
Fair |
HPMC: Hydroxy Propyl Methyl Cellulose,
PVP: polyvinyl pyrrolidone,
MCC: microcrystalline cellulose
EC: Ethyl Cellulose
*Coating I:mixture of methacrylic acid copolymer (L-100), triethyl citrate, titanium dioxide, and talc.
#Coating II:Mixture of methacrylic acid copolymer (S-100), titanium dioxide, talc and triacetin.
From above table observed that coating I, II, ethyl cellulose, magnesium stearate shows good fair flow property. PVP k-30, MCC has poor flow property. And HPMC K4M, METHOCEL has very very poor flow property.
C. Drug-excipients Compatibility studies:
As described in the methodology section the FT-IR studies were carried out for pure drug alone and along with polymers.The standard spectra of amoxicillin trihydrate shown in figure 5.137. The results are summarized as follows. An FT-IR spectrum of pure amoxicillin trihydrate is shown in the Figure 5.2. Similarly FT-IR spectra of amoxicillin trihydrate in combination with polymers are shown in Figures 5.3 to 5.6. These peaks were not affected and prominently observed in FT-IR spectra given in Figures 5.3 to 5.6. This indicates that there is no interaction between amoxicillin trihydrate and polymersand the drug was compatible with the formulation components.
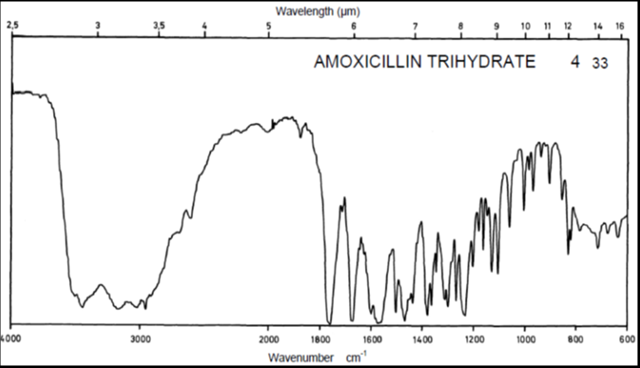
Figure 5.1: FTIR standard spectrum of amoxicillin trihydrate
Figure 5.2: FT-IR spectrum of pure amoxicillin trihydrate
Figure 5.3: FT-IR spectrum of physical mixture of amoxicillin trihydrate and other excipients
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Figure 5.4:FT-IR spectrum of physical mixture of amoxicillin trihydrate and ethyl cellulose
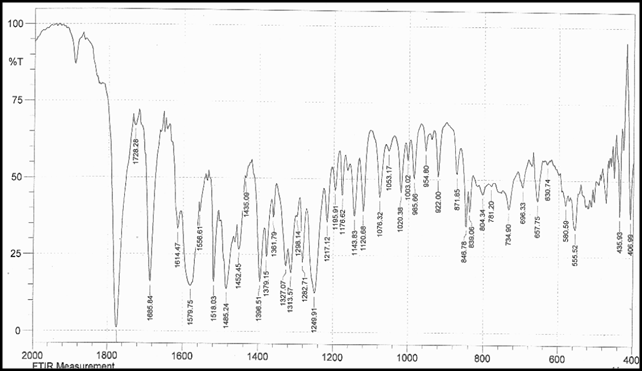
Figure 5.5: FT-IR spectrum of physical mixture of amoxicillin trihydrate and HPMC K4M
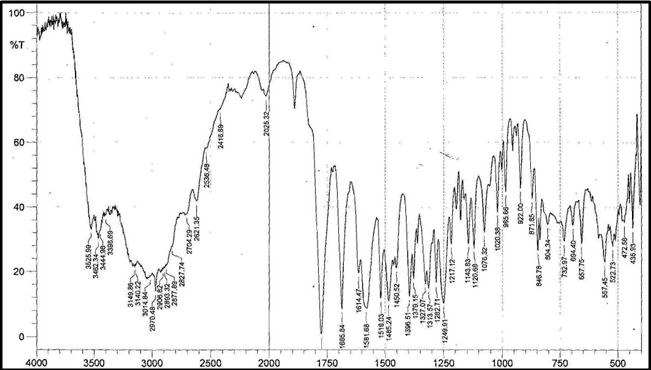
Figure 5.6: FT-IR spectrum of physical mixture of amoxicillin trihydrate and METHOCEL
FORMULATION DEVELOPMENT
Theoretical Drug Release Profile
The theoretical drug release profile calculation is important to evaluate the formulation with respect to release rates and to ascertain whether it releases the drug in a predetermined manner.Theoretically drug release profile was calculated by using the pharmacokinetic data. Attempts were made to develop formulations using available polymers, the combination of which shows dissolution profile that matches with the theoretical drug release profile.
Dose calculations: The total dose of amoxicillin for once daily modified release formulation was calculated by the following equation using the available pharmacokinetic data.26, 27, 28
F = bioavailability =85 to 90%
Css = steady state concentration = 2 µg/ml
Vd = volume of distribution = 0.3 l/kg = 18000ml per 60 kg.
T1/2 = half life = 1.02 hr
ClT = total clearance
XL = loading dose
DM = maintenance dose
DT = total dose = XL + DM
XL = Css* Vd
----------
F
= 2* 18000 / 0.9
= 40,000 µg = 40 mg
ClT = 0.693 Vd / T1/2 = 13860 ml/hr
DM = Css * ClT* Time
------------------
F
= 2 * 13860 *24 / 0.9 = 739.2 mg
DT = XL + DM = 40+739.2 = 779.2 mg
According to theoretical release pattern, once daily amoxicillin modified release formulation should release 40 mg in 1 hour, 30.8 mg every hour, and 100% in 24 hours.
Table 5.4: Dissolution profile of amoxicillin trihydrate according to theoretical calculations
|
Sampling time (hr) |
% drug release |
|
1 |
9.08 |
|
2 |
13.03 |
|
4 |
20.94 |
|
6 |
28.85 |
|
8 |
36.75 |
|
10 |
44.66 |
|
12 |
52.56 |
|
16 |
68.37 |
|
20 |
84.18 |
|
24 |
100 |
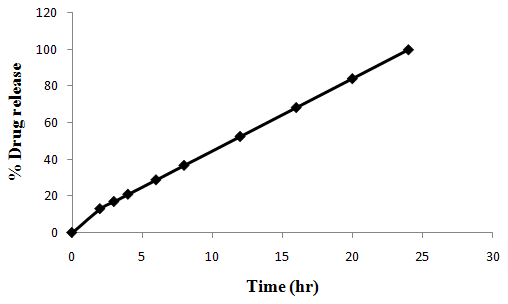
Figure 5.7:Dissolution profile of amoxicillin trihydrate according to theoretical calculation
TABLET
Development of the formulation in the present study was mainly based on the type of polymer, concentration of polymers, and the drug. Various polymers and excipients in different combinations were used so as to get tablet with good physical properties and in vitro drug release from tablet. Amoxicillin trihydrate is water soluble drug, and dose taken is 775 mg. So, in the present study attempts were made to get good physical and release profile of the tablets.
Manufacture of Tablets
As described in the methodology chapter the tablets were prepared by different method. In this method, all the excipients and drug were geometrically mixed and that blend was used for compression. Different polymers were used in different concentrations to get good release of drug. Different excipients like delayed release polymer, coating material, lubricants were used to get the tablets with acceptable physical properties.
Tablet prepared by using coating material and HPMC K4M:
The tablets were manufactured for the batches T1 to T3 using the ingredients shown in the Table 4.10. After getting all physical properties of the granule satisfactory, tablets were prepared. Physical as well as drug release pattern were studied for each batch. Physical properties of the granules are shown in theTable 5.5.The properties of the manufactured tablets areshown in theTable 5.6.
Table 5.5:Physical properties of the granules of the batches T1 to T3
|
Parameter |
T1 |
T2 |
T3 |
|
angle of repose(0) |
36.02 |
36.52 |
33.6 |
|
Bulk density(gm/ml) |
0.566 |
0.543 |
0.50 |
|
Tapped density(gm/ml) |
0.610 |
0.636 |
0.58 |
|
Haussner’s Ratio |
1.07 |
0.85 |
1.16 |
|
Compressibility Index (%) |
7.21 |
14.62 |
13.79 |
Table 5.6: Properties of the tablets for the batches T1 to T3
|
Parameter |
T1 |
T2 |
T3 |
|
Hardness (kg/cm2)(n=6) |
9.5 +0.9 |
9 + 0.2 |
12 + 0.5 |
|
Friability (%) |
0.0032 |
0.003 |
0.001 |
|
Uniformity of weight (gm) |
1.0687 +5% |
1.212 + 5% |
1.300 + 5% |
|
Drug content (%) (n=3) |
97.5+ 0.02 |
97.17+0.06 |
98.1+0.20 |
|
Thickness (mm)(n=6) |
6.79 +0.008 |
7.74 + 0.01 |
8.30 +0.02 |
Dissolution profile of the tablets for the batches T1 to T3:
After getting all the physical parameters satisfactory for batches T1 to T3, the dissolution of these batches was tested. Dissolution was carried out as per the procedure mentioned in methodology chapter. The details of the dissolution study for the tablets of the batches T1 to T3are given in the Table 5.7 and in Figure 5.8.
Table 5.7: In vitro release of Amoxicillin trihydrate from tablets of batch T1 to T3
|
Time (hours) |
Cumulative percentage of drug released (%) |
||
|
T1 (n=6) |
T2 (n=6) |
T3 (n=6) |
|
|
1 |
74.79+ 0.1 |
7.28+ 0.02 |
71.64+ 0.42 |
|
2 |
79.79+ 0.1 |
11.95+ 0.1 |
85.21+ 0.63 |
|
4 |
97.62+ 0.07 |
24.90+ 0.05 |
98.10+ 0.37 |
|
6 |
- |
30.34+ 0.1 |
- |
|
8 |
- |
34.77 + 0.2 |
- |
|
10 |
- |
55.81+ 0.08 |
- |
|
12 |
- |
62.63+ 0.3 |
- |
|
16 |
- |
82.39+ 0.36 |
- |
|
20 |
- |
98.03+ 0.2 |
- |
|
24 |
- |
- |
- |
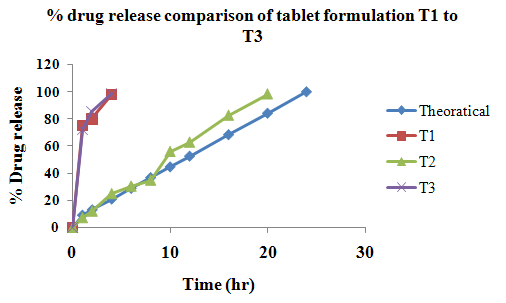
Figure 5.8:Comparison of in vitro release of Amoxicillin trihydrate from the tablets of the batches T1 to T3 with theoretical drug release profile.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Drug release profile of any of the formulations mentioned above did not appear to be closer to the theoretical drug release profile. As T1 showed 97.62 % in 4 hours, T2 showed 98.03% in 20 hours and 98.01 % in 4 hrs. This indicates that no formulation is showing acceptable drug release profile as theoretical profile. This could be because in T1 formulation batch there was only used coating material for drug release and no delayed release polymer used. So tablet had disintegrated quickly and drug release so fast. It could not be extend up to 24 hours. Batch formulation T2 made only by using delayed release polymer HPMC K4M and drug. These batch formed by direct compression method. The drug release found to be controlled in this formulation batch as compared to T1 formulation batch. But tablet thickness too much high than T1 formulation batch. If will adding more polymer for drug release extend up to 24 hours, then it will has more thickness and drug release also slow in initial hours as compared to theoretical drug release profile. So, this batch formulation modified by using coating material. Batch formulation T3, made by using delayed release polymer HPMC K4M and coating material. Coating of granule was made by dividing granules in three parts as formulation batch T1. But in this batch, drug release so quick due to fast disintegrate of tablet because of coating material.
However, this polymer used alone in three formulations could not control the drug release upto 24 hours and the aim was not achieved. Hence further attempts were made to improve the formulation.
Tablet prepared by using ethyl cellulose:
Polymer and coating material were used in the batches T4 to T7 to have an idea about the drug release. Based on the results obtained from the batches T1 to T3, the ethyl cellulose polymers were used in different proportions for the batches T4 to T7, so as to get the desired drug release profile. The tablets were manufactured for the batches T4 to T7 using the ingredients shown in the Table 4.10. After getting all physical properties of the granules satisfactory, tablets were prepared. Physical as well as drug release patterns were studied for each batch. Physical properties of the granules are shown in the Table 5.8. The properties of the manufactured tablets areshown in theTable 5.9.
Table 5.8:Physical properties of the granules of the batches T4 to T7
|
Parameter |
T4 |
T5 |
T6 |
T7 |
|
angle of repose(0) |
33.69 |
29.05 |
31.21 |
32.8 |
|
Bulk density(gm/ml) |
0.533 |
0.518 |
0.53 |
0.52 |
|
Tapped density(gm/ml) |
0.599 |
0.583 |
0.58 |
0.586 |
|
Haussner’s Ratio |
1.12 |
1.125 |
1.09 |
1.126 |
|
Compressibility Index (%) |
11.01 |
11.14 |
8.62 |
11.26 |
Table 5.9: Properties of the tablets for the batches T4 to T7
|
Parameter |
T4 |
T5 |
T6 |
T7 |
|
Hardness (kg/cm2)(n=6) |
10 + 0.2 |
9 +0.5 |
9 +0.5 |
9 +0.5 |
|
Friability (%) |
0.01 |
0.001 |
0.02 |
0.021 |
|
Uniformity of weight (gm) |
1.044 + 5% |
1.025 + 5% |
1.006 + 5% |
0.988 + 5% |
|
Drug content (%) (n=3) |
98.0+ 0.13 |
98.3+ 0.01 |
97.8+ 0.03 |
97.6+ 0.61 |
|
Thickness (mm)(n=6) |
7.12 + 0.02 |
6.31 + 0.01 |
6.31 +0.03 |
6.19 +0.02 |
Dissolution profile of the tablets for the batches T4 to T7
After getting all the physical parameters satisfactory for batches T4 to T7, the dissolution of these batches was tested. Dissolution was carried out as per the procedure mentioned in methodology chapter. The details of the dissolution study for the tablets of the batches T4 to T7 are given in the Table 5.10 and in Figure 5.9.
Table 5.10: In vitro release of Amoxicillin trihydrate from tablets of batch T4 to T7
|
Time (hours) |
Cumulative percentage of drug released (%) |
|||
|
T4 (n=6) |
T5 (n=6) |
T6 (n=6) |
T7 (n=6) |
|
|
1 |
6.44+ 0.02 |
6.50+ 0.01 |
7.02+ 0.83 |
7.96+ 0.12 |
|
2 |
8.30+ 0.37 |
9.90+ 0.38 |
10.11+ 0.64 |
11.08+ 0.34 |
|
4 |
10.75+ 0.64 |
12.38+ 0.72 |
14.37+ 0.27 |
15.40+ 0.56 |
|
6 |
16.70+ 0.29 |
17.69+ 0.64 |
19.83+ 0.81 |
20.38+ 0.82 |
|
8 |
19.60+ 0.37 |
20.71+ 0.38 |
21.75+ 0.93 |
23.27+ 0.71 |
|
10 |
23.67+ 0.08 |
25.53+ 0.18 |
27.39+ 0.67 |
29.84+ 0.93 |
|
12 |
27.14+ 0.62 |
28.27+ 0.92 |
30.00+ 0.30 |
32.19+ 0.41 |
|
16 |
32.70+ 0.11 |
34.62+ 0.15 |
36.12+ 0.53 |
38.30+ 0.34 |
|
20 |
38.27+ 0.27 |
40.36+ 0.25 |
44.50+ 0.41 |
49.74+ 0.50 |
|
24 |
48.89+ 0.33 |
50.42+ 0.74 |
55.68+ 0.63 |
57.20+ 0.09 |
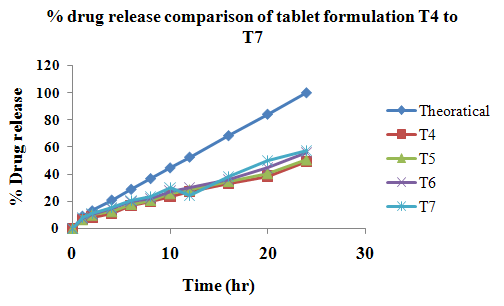
Figure 5.9:Comparison of in vitro release of Amoxicillin trihydrate from the tablets of the batches T4 to T7 with theoretical drug release profile.
Drug release profile of any of the formulations mentioned above did not appear to be closer to the theoretical drug release profile. As T4 showed 48.89% in 24 hours, T5 showed 50.42% in 24 hours, T6 showed 50.68% in 24 hours, T7 showed 57.20% in 24 hours. This indicates that no formulation is showing acceptable drug release profile as theoretical profile.
This could be because in T4 formulation batch there was used more concentration of polymer. So drug cannot release from tablet. So have to decrease this concentration for next batches.
So, next formulations of tablets were made by 6, 4, and 2% of ethyl cellulose polymer. But all formulation was failed to show drug release as compared to theoretical drug release profile.
However, this polymer used alone in four formulations could control the drug release in so much extend and the aim was not achieved. Hence further attempts were made to improve the formulation.
Tablet prepared by using ethyl cellulose and METHOCEL:
Individual polymer (HPMC K4M) was used in the batches T1 to T3 and ethyl cellulose in different concentration was used in the batches T4 to T7. But the selected polymer and in different concentration have proved to be unsuccessful. Therefore, based on the experience obtained with the first seven batches, further modifications in the formulations were attempted by the methocel polymers only i.e., by changing the proportion of combination of ethyl cellulose and methocel.
The tablets were manufactured for the batches T8 to T12 using the ingredients shown in the Table 4.10. After getting all physical properties of the granules satisfactory, tablets were prepared. Physical as well as drug release patterns were studied for each batch. Physical properties of the granules are shown in the Table 5.11. The properties of the manufactured tablets areshown in theTable 5.12.
Table 5.11:Physical properties of the granules of the batches T8 to T12
|
Parameter |
T8 |
T9 |
T10 |
T11 |
T12 |
|
angle of repose(0) |
34.55 |
33.62 |
30.32 |
28.89 |
35.72 |
|
Bulk density(gm/ml) |
0.666 |
0.4918 |
0.607 |
0.509 |
0.507 |
|
Tapped density(gm/ml) |
0.75 |
0.600 |
0.714 |
0.575 |
0.599 |
|
Haussner’s Ratio |
1.125 |
1.22 |
1.175 |
1.12 |
1.18 |
|
Compressibility Index (%) |
11.11 |
18.03 |
14.89 |
11.46 |
15.43 |
Table 5.12: Properties of the tablets for the batches T8 to T12
|
Parameter |
T8 |
T9 |
T10 |
T11 |
T12 |
|
Hardness (kg/cm2) (n=6) |
10 + 0.2 |
9 +0.4 |
9.5 +0.5 |
10 + 0.2 |
9.5 + 0.3 |
|
Friability (%) |
0.03 |
0.02 |
0.005 |
0.01 |
0.002 |
|
Uniformity of weight (gm) |
0.99 +5% |
0.99 + 5% |
0.99 +5% |
0.99 +5% |
0.99 + 5% |
|
Drug content (%) (n=3) |
97.36 + 0.10 |
96.87 + 0.08 |
98.01+0.23 |
97.74+0.17 |
96.97 + 0.25 |
|
Thickness (mm) (n=6) |
6.71 + 0.02 |
6.68 + 0.03 |
6.75 + 0.01 |
6.68 + 0.03 |
6.80 + 0.02 |
Dissolution profile of the tablets for the batches T8 to T12
After getting all the physical parameters satisfactory for batches T8 to T12, the dissolution of these batches was tested. Dissolution was carried out as per the procedure mentioned in methodology chapter. The details of the dissolution study for the tablets of the batches T8 to T12 are given in the Table 5.13 and in Figure 5.10.
Table 5.13: In vitro release of Amoxicillin trihydrate from tablets of batch T8 to T12
|
Time (hrs) |
Cumulative percentage of drug released (%) |
||||
|
T8 (n=6) |
T9 (n=6) |
T10 (n=6) |
T11 (n=6) |
T12 (n=6) |
|
|
1 |
8.02+ 0.23 |
8.23+ 0.17 |
8.7+ 0.83 |
8.9+ 0.27 |
9.02+ 0.29 |
|
2 |
11.2+ 0.51 |
11.0+ 0.72 |
11.2+ 0.72 |
11.83+ 0.20 |
12.69+ 0.31 |
|
4 |
16.11+ 0.78 |
17.03+ 0.44 |
17.0+ 0.66 |
18.44+ 0.82 |
19.66+ 0.75 |
|
6 |
22.0+ 0.37 |
24.22+ 0.38 |
25.97+ 0.28 |
26.38+ 0.79 |
26.87+ 0.88 |
|
8 |
25.74+ 0.32 |
27.23+ 0.91 |
30.01+ 0.85 |
32.81+ 0.27 |
35.72+ 0.47 |
|
10 |
30.38+ 0.78 |
34.41+ 0.19 |
37.82+ 0.61 |
39.30+ 0.31 |
42.77+ 0.22 |
|
12 |
35.42+ 0.38 |
38.91+ 0.24 |
43.55+ 0.23 |
47.11+ 0.43 |
49.83+ 0.02 |
|
16 |
41.51+ 0.76 |
48.05+ 0.11 |
52.20+ 0.56 |
59.36+ 0.57 |
64.90+ 0.13 |
|
20 |
53.62+ 0.81 |
62.88+ 0.09 |
69.74+ 0.78 |
73.94+ 0.69 |
79.22+ 0.83 |
|
24 |
61.79+ 0.56 |
70.29+ 0.02 |
78.00+ 0.01 |
84.03+ 0.53 |
89.31+ 0.02 |
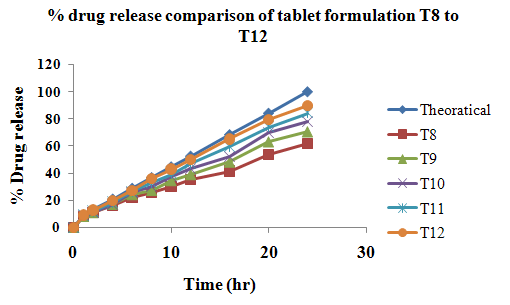
Figure 5.10:Comparison of in vitro release of Amoxicillin trihydrate from the tablets of the batches T8 to T12 with theoretical drug release profile.
Formulation T8 showed 61.79% in 24 hours, T9 showed 70.29% in 24 hours, T10 showed 78.0% in 24 hours, T11 showed 84.03% in 24 hours and T12 showed 89.31% in 24 hours.
Early formulation of batches T4 to T7 was made by using ethyl cellulose polymer for control drug release from tablet. But only ethyl cellulose even in low concentration drug release found to be very much slow.so, have to add methocel to that formulation. So, now in tablet formulation batches T8 to T12, there was different proportion of the ethyl cellulose and methocel.
In T8 tablet formulation, drug release was found to be more than early single polymer used formulation. But not near to the theoretical drug release profile. So again increase the methocel amount in next formulation.
Different formulation was made by using different proportion of two polymers. Drug release found to be near to the theoretical drug release profile. But still have to increase drug release from tablet. Hence further attempts were made to improve the formulation.
Tablet prepared by using ethyl cellulose and methocel with different hardness of tablet:
Individual polymer (HPMC K4M) was used in the batches T1 to T3 and ethyl cellulose in different concentration was used in the batches T4 to T7. But the selected polymer and in different concentration have proved to be unsuccessful. Therefore, based on the experience obtained with the first seven batches, further modifications in the formulations were attempted by the methocel polymers only i.e., tablets were manufactured for the batches T8 to T12 by changing the proportion of combination of ethyl cellulose and methocel. Further change the proportions with comparison of hardness of tablet.
The tablet was manufactured for the batches T13 using the ingredients shown in the Table 4.10 with different hardness. After getting all physical properties of the granules satisfactory, tablets were prepared. Physical as well as drug release patterns were studied for each batch. Physical properties of the granules are shown in the Table 5.14. The properties of the manufactured tablets areshown in theTable 5.15.
Table 5.14:Physical properties of the granules of the batches T13
|
Parameter |
T13 |
|
angle of repose(0) |
31.83 |
|
Bulk density(gm/ml) |
0.546 |
|
Tapped density(gm/ml) |
0.612 |
|
Haussner’s Ratio |
1.11 |
|
Compressibility Index (%) |
10.70 |
Table 5.15: Properties of the tablets for the batches T13 and T13.1
|
Parameter |
T13 |
T13.1 |
|
Hardness (kg/cm2)(n=6) |
10 + 0.3 |
6.5 + 0.1 |
|
Friability (%) |
0.07 |
0.005 |
|
Uniformity of weight (gm) |
0.99 + 5.0% |
0.99 + 5.0% |
|
Drug content (%) (n=3) |
97.88 + 0.06 |
98.03 + 0.03 |
|
Thickness (mm)(n=6) |
6.82 + 0.02 |
6.62 + 0.02 |
Dissolution profile of the tablets for the batches T13 and T13.1
After getting all the physical parameters satisfactory for batches T13 and T13.1, the dissolution of these batches was tested. Dissolution was carried out as per the procedure mentioned in methodology chapter. The details of the dissolution study for the tablets of the batches T13 and T13.1 are given in the Table 5.16 and in Figure 5.11.
Table 5.16: In vitro release of Amoxicillin trihydrate from tablets of batch T13 and T13.1
|
Time (hours) |
Cumulative percentage of drug released (%) |
|
|
T13 (n=6) |
T13.1 (n=6) |
|
|
1 |
9.0+ 0.25 |
9.1+ 0.72 |
|
2 |
13.02+ 0.78 |
14.0+ 0.61 |
|
4 |
19.89+ 0.51 |
20.4+ 0.44 |
|
6 |
27.12+ 0.62 |
27.99+ 0.19 |
|
8 |
36.01+ 0.94 |
37.01+ 0.15 |
|
10 |
43.1+ 0.48 |
44.0+ 0.28 |
|
12 |
50.37+ 0.75 |
51.99+ 0.64 |
|
16 |
66.40+ 0.82 |
67.89+ 0.82 |
|
20 |
80.08+ 0.38 |
82.78+ 0.91 |
|
24 |
92.45+ 0.71 |
97.72+ 0.37 |
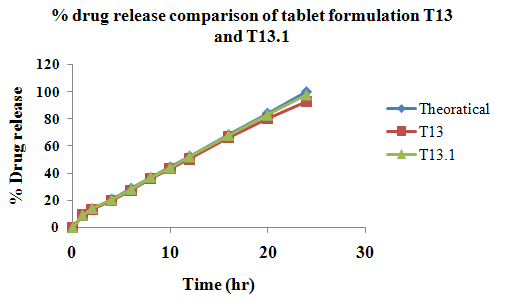
Figure 5.11:Comparison of in vitro release of Amoxicillin trihydrate from the tablets of the batches T13 and T13.1 with theoretical drug release profile.
Formulation T13 and T13.1 drug release was found to be 92.45% and 97.72% in 24 hours. In formulation T13, the drug release was found to be near to theoretical drug release profile. But drug release at 24 hour was little less. If further change the proportion and increase the amount of methocel then the drug release not in control up to 24 hours. Hence, there was no further modification of proportions and next formulation made by only decrease the hardness and showed that there was increase in the drug release.
Formulation T13.1 is the successful formulation which drug release close to the theoretical drug release profile. But further it needs to confirm.
Comparison of f1andf2values for the tablets T13.1 with theoretical drug release profile:
The suitability of the formulation was further confirmed by comparing the dissolution data with that of the theoretical drug release profiles. The similarity (f2value) and dissimilarity (f1value) factors of the batch T13.1 compared to the theoretical drug release profile are given in the Table 5.17. Perusal to the Table 5.17 indicates that formulation T13.1,f1 and f2values for the formulation are falling within the range.Further it confirmed by the reproducible batch of formulation T13.1.
Table 5.17: Comparison of f1andf2values for the tablets of the batch T13.1 with theoretical drug release profile
|
Formulation |
f2value (Similarity factor) |
Acceptable range |
f1value (Dissimilarity factor) |
Acceptable range |
|
T13.1 |
92.37 |
50-100 |
1.75 |
0-15 |
Reproducibility of the Batch T13.1
Batch to batch uniformity is very much essential for obtaining reproducible results. In order to verify this, the tablets of the batch T13.1 were manufactured for the second time. Release studies were conducted as specified for in vitro study.
The release patterns of the first and second trials are shown in the Figure 5.12. The perusal to the Figure 5.12 indicates that, the graphs are overlapping proving the reproducibility. Data for the in vitrorelease of amoxicillin trihydrate from the tablets of the batch T13.1 of first and second (reproducible batch) trails are shown in the Table 5.18.
Table 5.18: Data for the in vitrorelease of Amoxicillin trihydrate from the tablets of the batch T13.1 of first and second (reproducible batch) trails
|
Time (hours) |
Cumulative percentage of drug released (%) |
|
|
First trial |
Second trial |
|
|
1 |
9.1+ 0.72 |
9.22+ 0.18 |
|
2 |
14.0+ 0.61 |
14.26+ 0.39 |
|
4 |
20.4+ 0.44 |
21.81+ 0.61 |
|
6 |
27.99+ 0.19 |
27.00+ 0.77 |
|
8 |
37.01+ 0.15 |
39.73+ 0.83 |
|
10 |
44.0+ 0.28 |
44.27+ 0.55 |
|
12 |
51.99+ 0.64 |
53.02+ 0.71 |
|
16 |
67.89+ 0.82 |
68.90+ 0.09 |
|
20 |
82.78+ 0.91 |
83.22+ 0.27 |
|
24 |
97.72+ 0.37 |
96.92+ 0.02 |
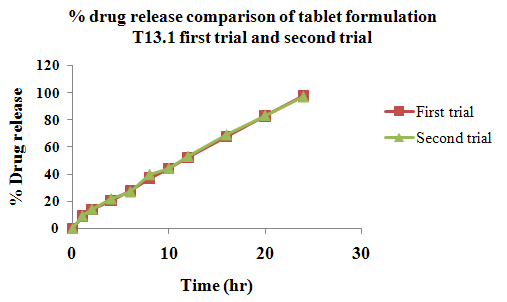
Figure 5.12: In vitrorelease of Amoxicillin trihydrate from the tablets of the batch T13.1 of firstand second (reproducible) trials
Comparison of f1andf2values for the tablets of first and second trials of batches of T13.1
The similarity (f2value) and dissimilarity (f1value) factors of the batch T13.1 compared to the theoretical drug release profile are given in the Table 5.19. Perusal to the Table 5.19 indicates that formulation is showing the values within the acceptable range.
Table 5.19: Comparison of f1andf2values for the tablets of the batches T13.1
|
Formulation |
f2value (Similarity factor) |
Acceptable range |
f1value (Dissimilarity factor) |
Acceptable range |
|
T13.1 |
90.75 |
50-100 |
1.97 |
0-15 |
Kinetics of In Vitro Drug Release
The dosage forms most commonly release the drug either in the zero order or in the first order pattern. Modified release dosage forms of Amoxicillin trihydrate were prepared and studied for their dissolution behavior. In vitrorelease data of time points between 1 to 24 hours were considered and treated for following kinetic principles and values are shown in Table 5.20. The release profiles of amoxicillin trihydrate from the tablets of the formulation T13.1 were processed into graphs (Figures 5.13 and 5.14) for comparison of different orders of drug release and to understand the linear relationship, i.e., kinetic principles. The data were processed for regression analysis using MS-Excel statistical functions. The equations for the line and regression coefficient are shown in the corresponding figures and comparison is made in the Table 5.21.
Table 5.20:In vitrorelease of amoxicillin trihydrate from the tablets of T13.1
|
Time (hours) |
% drug released |
Log % drug released |
% drug unreleased |
Log % drug unreleased |
|
1 |
9.1 |
0.9590 |
90.9 |
1.9585 |
|
2 |
14.0 |
1.1461 |
86.0 |
1.9344 |
|
4 |
20.4 |
1.3096 |
79.6 |
1.9000 |
|
6 |
27.99 |
1.4470 |
72.01 |
1.8573 |
|
8 |
37.01 |
1.5683 |
62.99 |
1.7992 |
|
10 |
44.0 |
1.6434 |
56.0 |
1.7481 |
|
12 |
51.99 |
1.7159 |
48.01 |
1.6813 |
|
16 |
67.89 |
1.8318 |
32.11 |
1.5066 |
|
20 |
82.78 |
1.9179 |
17.22 |
1.2360 |
|
24 |
97.72 |
1.9899 |
2.28 |
0.3579 |
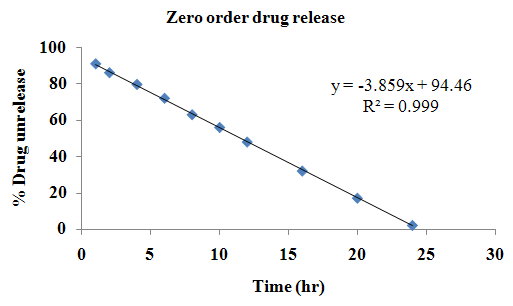
Figure 5.13:In vitrorelease profile of Amoxicillin trihydrate from tablets of T13.1 fitted in zero order release
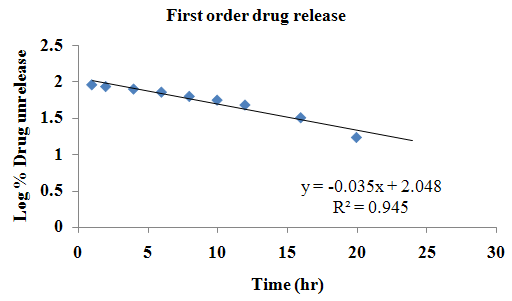
Figure 5.14:In vitrorelease profile of Amoxicillin trihydrate from tablets of T13.1 fitted in first order release
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table 5.21: Comparison of orders of in vitro release of amoxicillin trihydrate from tablets of T13.1
|
Formulation |
Regression equations |
|
|
|
Zero order |
First order |
|
T13.1 |
y = -3.8595x + 94.464 |
log y = -0.0356x + 2.0486 |
Perusal to theFigures 5.13 and 5.14reveals that the formulations did not follow a First-order release pattern but followed the Zero-order equation. The formulation showed a fair linearity, with regression value of 0.9997.
Release Mechanisms
To study the release mechanisms of amoxicillin trihydrate the data of in vitro drug release was verified using Higuchi’s model, Korsmeyer-Peppas model, and Hixon-Crowell cube root law models.
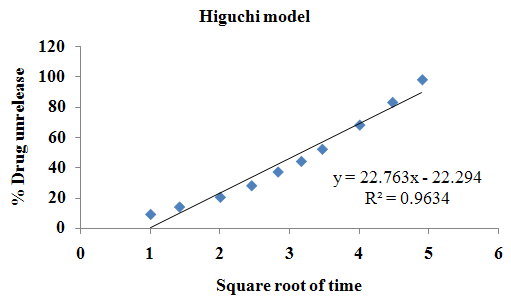
Figure 5.15:In vitrorelease profile amoxicillin trihydrate from tablets of T13.1 fitted in Higuchi’s Plot
Table5.22: Fitting of the Hixon-Crowell cube root law for in vitro release of amoxicillin trihydrate from the tablets of T13.1
|
Time (hour) |
M01/3-M1/3 |
|
1 |
0.291 |
|
2 |
0.456 |
|
4 |
0.682 |
|
6 |
0.967 |
|
8 |
1.333 |
|
10 |
1.643 |
|
12 |
2.032 |
|
16 |
2.966 |
|
20 |
4.217 |
|
24 |
7.785 |
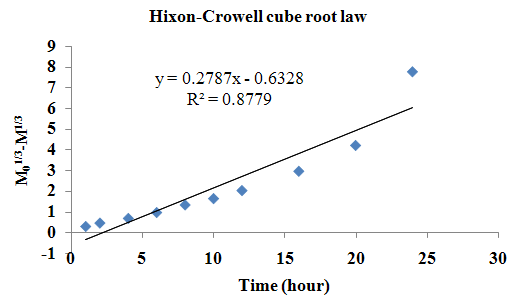
Figure 5.16: Fitting of the Hixon-Crowell cube root law for in vitro release of amoxicillin trihydrate from the tablets of T13.1
In order to explore more precise mechanism of release of amoxicillin trihydrate from in house developed tablets, the dissolution data was also fitted to the well known exponential equation (Korsmeyer equation) as shown in the Figure 5.17, which is often used to describe the drug release behavior from polymeric systems.
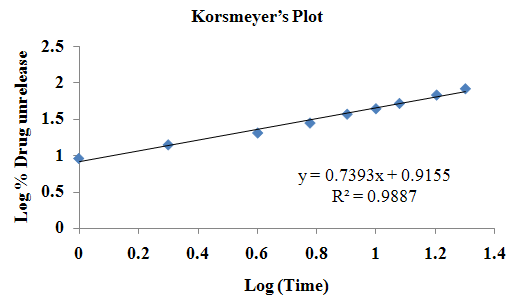
Figure 5.17: In vitrorelease profile of amoxicillin trihydrate from the tablets of T13.1 fitted in Korsmeyer’s Plot.
Table5.23: Regression equations of in vitro release of Amoxicillin trihydrate from the tablets of T13.1
|
Formulation T13.1 |
In vitrorelease of amoxicillin trihydrate from the tablets of T13.1 |
||
|
Higuchi’s model
|
Hixon-Crowel model |
Korsmeyer Peppas model |
|
|
|
y = 22.76x - 22.294 R² = 0.9634 |
y = 0.2787x -0.632 R² = 0.8779 |
y = 0.7393x +0.9155 R² =0.9887
|
Application of Hixon–Crowell cube root law, the equation (M01/3 – M1/3) = kt, provides information about the release mechanism, namely dissolution rate limited. Application of Higuchi’s equation (M = K t1/2) provides information about the release mechanism, namely diffusion rate limited. Korsmeyer-Peppas model indicates that release mechanism is not well known or more than one type of release phenomena could be involved. The 'n' value could be used to characterize different release mechanisms as:
Table5.24: Slope of Korsmeyer-Peppas equation and proposed release mechanisms
|
Slope ( n ) |
Mechanism |
|
<0.5 |
Fickian diffusion (Higuchi Matrix) |
|
0.5 <n< 1 |
Non-Fickian diffusion |
|
1 |
Case II transport |
The data of average values were processed as per Hixon-Crowell cube root law and are given in the Tables 5.22 and the Figure 5.16. The data of average values were processed as per Higuchi’s equation and are represented in the Figure 5.15. The data of average values were processed as per Korsmeyer-Peppas model and are represented in the Figure 5.17. The linearity of data for all the models was identified from the Figures. The equations were generated through statistical procedures and reported in the Table 5.23.
Perusal to the Table 5.23 indicates that R2 values are higher for the Higuchi’s model compared to Hixon – Crowell for the tablets of T13.1. Hence amoxicillin trihydrate release from the tablets of T13.1 followed diffusion rate controlled mechanism.
According to Korsmeyer-Peppas model,a value of slope is between 0.5 to 1.0 indicates the Non-Fickian diffusion and hence release mechanism from tablets of T13.1follows Non-Fickian diffusion.
Accelerated stability study
Stability of a drug has been defined as the ability of a particular formulation, in a specific container, to remain within its physical, chemical, therapeutic, and toxicological specifications.
Tablets of T13.1 were kept for accelerated stability study at 40 + 2oC and 75 + 5% RH for 1 month in the modified stability chamber.
After a period of one month, the samples were observed for any change in physical parameters. It was observed that surface was devoid of any change in color or appearance of any kind of spots on it. It was also noted that surface was free of any kind of microbial or fungal growth or bad odor. No changes in the smoothness of the tablets were noted. The drug content of the formulations was found 98.03% for tablets of T13.1, which shows that, there is no decrease in drug content and difference is insignificant. The in vitro release of the samples after one month storage compared with release profile of sample at zero are shown in the Table 6.25 for T13.1. Graphical representations are shown in Figure5.18.
Table 5.25: In Vitrorelease of amoxicillin trihydrate from tablets of T13.1 on zero day and samples after one month accelerated stability studies
|
Time (hours) |
Cumulative percentage of drug released (%) |
|
|
Tablets of zero day |
Tablets after one month |
|
|
1 |
9.1+ 0.72 |
8.78+ 0.65 |
|
2 |
14.0+ 0.61 |
13.29+ 0.84 |
|
4 |
20.4+ 0.44 |
21.00+ 0.28 |
|
6 |
27.99+ 0.19 |
26.32+ 0.31 |
|
8 |
37.01+ 0.15 |
38.23+ 0.11 |
|
10 |
44.0+ 0.28 |
43.84+ 0.77 |
|
12 |
51.99+ 0.64 |
52.00+ 0.83 |
|
16 |
67.89+ 0.82 |
66.27+ 0.64 |
|
20 |
82.78+ 0.91 |
81.99+ 0.51 |
|
24 |
97.72+ 0.37 |
96.04+ 0.80 |

Figure 5.18: In vitrorelease of amoxicillin trihydrate tablets from T13.1 on zero day and after one month of accelerated stability studies
By comparison, it was found that after a period of one month of storage there were no changes in the physical as well as drug release profiles of the tablets of both the batches and both were imitating the same drug release pattern. The f1 and f2 values in the comparison of release before and after one-month storage (at accelerated conditions) were found 1.79 and 91.76 respectively for tablet (T13.1).
CAPSULE
Development of the formulation in the present study was mainly based on the type of polymer, concentration of polymers, and the drug. Various polymers and excipients in different combinations were used so as to get desire in vitro drug release from tablet. Amoxicillin trihydrate is water soluble drug, and dose taken is 775 mg. So, in the present study attempts were made to get capsule with desire drug release profile of the capsule.
Manufacture of capsule
As described in the methodology chapter the, Granules prepared by wet granulation method by mixing ingredients given in the table 4.11, compress it in tablet form with higher hardness. Crush tablet and pass it from sieve 14#. Fill it in a 00size hard gelatin capsule shell by using hand operated capsule filling machine.Different polymers were used in different concentrations to get good release of drug. Different excipients like delayed release polymer, coating material, lubricants were used to get good drug release.
Table 5.26: Uniformity of weight and drug content of capsule formulations C1 to C6
|
Formulations |
Uniformity of weight of net content(gm) |
Drug content (%) (n=3) |
|
C1 |
1.065+ 7.5% |
97.02+ 0.52 |
|
C2 |
1.212+ 7.5% |
97.32+ 0.21 |
|
C3 |
1.300+ 7.5% |
98.00+ 0.11 |
|
C4 |
1.044+ 7.5% |
97.98+ 0.73 |
|
C5 |
1.054+ 7.5% |
97.99+ 0.85 |
|
C6 |
1.10+ 7.5% |
98.76+ 0.29 |
Dissolution profile of the capsules for the batches C1 to C6
After getting uniformity of weight of net content and drug content satisfactory for batches C1 to C6, the dissolution of these batches was tested. Dissolution was carried out as per the procedure mentioned in methodology chapter. The details of the dissolution study for the capsules of the batches C1 to C6 are given in the Table 5.27 and in Figure 5.19.
Table 5.27: In vitro release of Amoxicillin trihydrate from capsules of batch C1 to C6
|
Time (hrs) |
Cumulative percentage of drug released (%) |
|||||
|
C1 (n=6) |
C2 (n=6) |
C3 (n=6) |
C4 (n=6) |
C5 (n=6) |
C6 (n=6) |
|
|
1 |
92.71+ 0.61 |
94.03+ 0.52 |
83.00+ 0.32 |
36.08+ 0.11 |
25.23+ 0.30 |
20.63+ 0.28 |
|
2 |
- |
- |
97.01+ 0.67 |
67.91+ 0.38 |
38.34+ 0.21 |
31.28+ 0.15 |
|
4 |
- |
- |
- |
96.45+ 0.63 |
49.65+ 0.66 |
45.37+ 0.62 |
|
6 |
- |
- |
- |
- |
62.88+ 0.82 |
53.45+ 0.85 |
|
8 |
- |
- |
- |
- |
78.62+ 0.62 |
66.27+ 0.49 |
|
10 |
- |
- |
- |
- |
86.13+ 0.44 |
71.83+ 0.64 |
|
12 |
- |
- |
- |
- |
97.24+ 0.17 |
84.00+ 0.39 |
|
16 |
- |
- |
- |
- |
- |
98.11+ 0.13 |
|
20 |
- |
- |
- |
- |
- |
- |
|
24 |
- |
- |
- |
- |
- |
- |
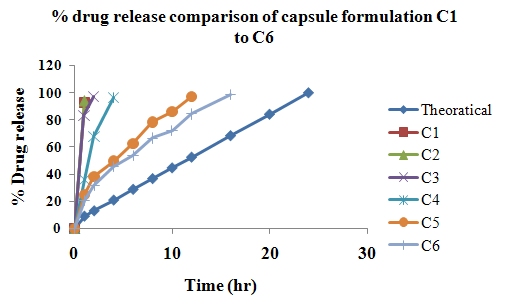
Figure 5.19:Comparison of in vitro release of Amoxicillin trihydrate from the capsules of the batches C1 to C6 with theoretical drug release profile.
Drug release profile of any of the formulations mentioned above did not appear to be closer to the theoretical drug release profile. As C1 showed 92.71 % in 1 hour, C2 showed 94.03% in 1hours, C3 showed 97.01 % in 2 hours, C4 showed 96.45 % in 4 hours, C5 showed 97.24 % in 12hours, C6 showed 98.11 % in 16hours. This indicates that no formulation is showing acceptable drug release profile as theoretical profile. This could be because in C1 formulation batch there was only used coating material for drug release and no delayed release polymer used. So granule was quickly dissolved and drug release so fast. It could not be extend up to 24 hours. Batch formulation C2 made only by using delayed release polymer HPMC K4M and drug. But it also could not be extend up to 24 hours. In formulation C3, coating is only more thing than the C2 showed no more drug release extend. In formulation C4 there was using directly high concentration of polymer i.e. 8% ethyl cellulose. But it also fails to extend drug release. In C5 formulation, there was 15% ethyl cellulose polymer used. But it only extends drug release up to 12hours. Finally in C6 formulation, there was 20% polymer used. But drug release only extends up to 16hours. However it was more than C5 formulation. But initial drug release was found to be more as compared to theoretical drug release profile. And if again add more concentration of the polymer then it can not fill in to even two 00size of capsule.
So, all formulation fails to match drug release with theoretical drug release profile. Capsule formulation was found to be not optimized. Capsule formulations not extend up to 24hrs drug release, so it not needs to compare with the tablet formulation.
Review of Literature
The chapter “Literature Review” contained review on drug and excipients. Extensive literature survey was done on dosage forms of amoxicillin trihydrate for selection of polymer, excipients, and method of manufacturing. A detailed description about amoxicillin trihydrate, polymer, and excipients were discussed.
Methodology
In order to solve the objectives of this work, suitable analytical method (UV Spectroscopy) was established and validated in distilled water, 50mM of potassium phosphate monobasic buffer at pH 4, 6 and 6.8 buffer solutions. Physical properties of Amoxicillin trihydrate and polymers like loose bulk density, tapped bulk density, compressibility index and angle of repose were determined. Formulations for amoxicillin trihydrate 775mg tablets and capsules were developed and were evaluated for pharmacopoeial and nonpharmacopoeial (industry specified) tests. Polymer and concentration of polymer were optimized for tablets. And it also tried for the capsule dosage forms. Tablet and capsules were prepared with different polymers like HPMC (K4M), ethyl cellulose, methocel, and coating material in order to optimize one final formula for dosage form. Tablets were evaluated for physical and chemical properties. For tablets in vitro release was carried out in distilled water for 24 hours using USP type II apparatus at 100 rpm. Capsule evaluation like weight uniformity, drug content and in vitro drug release were also done. Short term accelerated stability study of optimized formulations of amoxicillin trihydrate 775mg tablets were carried out at 40 + 2 oC and 75 + 5% RH for one month.
Results and discussion
The results and discussion of different methods of this thesis were described under different headings using graphs and tables. No interference due to additives in the estimation of amoxicillin trihydrate was observed (Figures 5.1 to 5.6). Different polymers were used to get a suitable formulation of tablets and capsule and finally were optimized to get optimized formulations. The optimized formulation consists of methocel and ethyl cellulose polymer. Tablets were evaluated for pharmacopoeial and nonpharmacopoeial (industry specified) tests and were found to be within the prescribed limits. Amoxicillin trihydrate tablets were prepared with different formulae for optimized formulations that show f1and f2in prescribed limits when using theoretical drug release profile as reference standards. The average f1 and f2 values of optimized formulation were found to be 1.75 and 92.37 for tablets respectively. The R2 = 0.9997 (figure 5.13) for the zero order release and for first order release R2 = 0.9451 (figure 5.14). Hence the release of amoxicillin trihydrate from developed formulations was considered to be zero order. The Higuchi’s equation showed R2 = 0.9634 (figure 5.15) and also when the data was fitted in to Korsmeyer et al equation it showed R2 = 0.9887 (figure 5.17) with slope (n) value of 0.7393 which is between 0.5 and 1.0. Thus, diffusion of the drug was the main mechanism for drug release for the optimized formulation. Formulation was found to be reproducible and stable for one month under accelerated stability condition. Capsule evaluation of weight uniformity and drug content was found to be passed but it fails in in vitro drug release test. And drug release found to be extending only up to 16hrs not up to 24hrs. So capsule was found to be not optimized formula.
Conclusions
The conclusions drawn from the present investigation were given below;
1. Suitable analytical method based on UV-Visible spectrophotometer was developed for amoxicillin trihydrate. ?maxof 272.6nm, 272.2nm, 272.8nm and 272.6nm nm and were identified in distilled water, 50mM potassium phosphate monobasic buffer at 4 pH, at 6 pH solution and at 6.8 pH buffer solution respectively.
2. From the FT-IR spectra the interference was verified and found that amoxicillin trihydrate did not interfere with the excipients used.
3. Procedure to manufacture tablets and capsules by different method was established.
4. Tablets of amoxicillin trihydrate (T13.1) were successfully prepared using ethyl cellulose (50%) and methocel (50%) by wet granulation method.
5. The tablets were evaluated for pharmacopoeial and non-pharmacopoeial (industry specified) tests. Based on the results, T13.1 was identified as better formulation amongst all formulations developed for tablets.
6. Tablets of the formulation T13.1 passed all official and unofficial quality control tests.
7. In vitro release profiles of optimized formulations of amoxicillin trihydrate tablets (T13.1) were found to be similar to that of theoretical drug release profile. The f1 and f2 values for the comparison of release of drugs from the formulation T13.1 with the theoretical drug release profile were found to be 1.75 and 92.37respectively, for tablets.
8. The manufacturing procedure was standardized and found to be reproducible. The f1 and f2 values for the comparison of release of drugs of first and second (reproducible) trial were found to be1.97 and90.75respectively for the tablets of the formulation T13.1.
9. Amoxicillin trihydraterelease from the tablets of T13.1 formulation follows zero - order kinetics.
10. Amoxicillin trihydrate release from the tablets of T13.1 formulation follows Higuchi model.
11. Release mechanism of amoxicillin trihydrate from tablets of T13.1 formulation follows Non-Fickian diffusion.
12. After one month of accelerated stability studies developed formulation was found to be stable. The f1 and f2 values in the comparison of release before and after one-month storage (at accelerated conditions) were found to be1.79 and 91.76for the tablets of the formulation T13.1.
13. Capsules of amoxicillin trihydrate were prepared using ethyl celluloseand methocel by slugging method.
14. The capsules were evaluated for weight uniformity, content uniformity and in vitro drug release.
15. Capsules of the formulation were passed the weight uniformity and drug content test but fails to in vitro drug release test.
16. In vitro drug release of the capsule formulation C6 only up to 16hours not to 24hours.
17. In comparison of tablet and capsule formulation, tablet was found to be successful formulation as compared to tablet dosage form.
The conclusions arrived in this thesis indicated that the modified release tablet of amoxicillin trihydrate developed in this investigation releases drug equivalent to theoretical drug release, based on in vitro release studies.
Further studies are needed to investigate these formulations for its performance in vivo. Capsule formulation also tries by using different method like extrusion spheronisation method etc.
Thus the objectives of the present thesis are achieved.
The result of the study indicates that modified release tablets of amoxicillin trihydrate that can be successfully prepared.
Appendix A
List of abbreviations
API Active Pharmaceutical Ingredient
Avg. average
E.g. Example
EC Ethyl Cellulose
EP European Pharmacopoeia
ER Extended Release
f1 Difference factor
f2 Similarity factor
FT-IR Fourier transform infrared spectroscopy
GI Gastro Intestinal
GIT Gastro Intestinal Tract
HCl Hydrochloric acid
HPMC Hydroxy Propyl Methyl Cellulose
hrs Hours
ICH The international conference on harmonization of technical requirements for registration of pharmaceuticals for human use
IP Indian Pharmacopoeia
IR Immediate Release
LBD Loose Bulk Density
MCC Microcrystalline cellulose
MIC Minimal Inhibitory Concentration
MR Modified Release
PBP Penicillin Binding Protein
PVP Polyvinyl Pyrrolidone
RH Relative Humidity
rpm rotations per minute
RT Retention Time
SD Standard Deviation
Sec. Seconds
Std. Standard
TBD Tapped Bulk Density
UDP Uridinediphosphate
USP United State Pharmacopoeia
Wt. Weight
References:
1. The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit, London, 29 July 1999. ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003664.pdf
2. “Guidance for Industry SUPAC-MR: Modified Release Solid Oral Dosage Forms”, July-1998. fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070640.pdf
3. australianprescriber.com/magazine/22/4/88/90/ - Australia
4. Dr. Ali R Rajabi-Siahboomi, “An Over view of Current Oral Modified Release Technologies”, Drug Delivery Oral, business briefing: pharmatech, 2003, pp. 181-184.
5. Syed azeem, Shawetasharma, “Immediate Release Drug Delivery Systems: A Review”, International Journal of Biopharmaceutical & Toxicological Research, May 2011, pp. 24-25.
6. Madhusudhanpogula, S. Nazeer, “Extended Release Formulation”, International Journal of Pharmacy & Technology, Dec-2010, 2, pp. 625-84.
7. Araya Raiwa, , “Formulation Development Strategies For Oral Extended Release Dosage Forms”, Bangkok, Thailand, May, 2011.
8. Formulation, solid dosage form-tablet formulation.vinensia.com/2011/02/tablet-solid-dosage-form.html
9. K.Senthil Kumar, M.pharm thesis, “pharmaceutical technology-II, tablet”, QIS college of pharmacy- Ongole A.P, India.
10. Formulation, solid dosage form-capsule formulation.vinensia.com/2011/04/capsule-solid-dosage-form.html
11. Prof. James a. Syce, m.pharm thesis, “The formulation, manufacture and evaluation of capsules containing freeze-dried aqueous extracts of leonotisleonorusor menthalongifolia”, University of the Western Cape, South africa, June 2006.
12. medicalnewstoday.com/articles/10278.php
13. Antibiotic review, April 2004. faculty.smu.edu/jbuynak/Medicinal_Outline_11_4_04.pdf
14. emedicinehealth.com
15. Antibiotic guideline palmta.org/uploads/1325495522735463Antibiotic%20Guideline.pdf
16. “Guidance for Industry Streptococcal Pharyngitis and Tonsillitis - Developing Antimicrobial Drugs for Treatment”, july-1998. fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071182.pdf
17. ncbi.nlm.nih.gov
18. Practice Guidelines And Protocols, Student Medical Services and District Nursing Services, 3rd Edition, 2008, pp. 75-78.
19. Wexner medical center, US news and world report best hospital 2011. medicalcenter.osu.edu/patientcare/healthcare_services/lung_diseases/tonsils/Pages/index.aspx
20. SimarPreetKaur, Rekha Rao, Sanju Nanda, “Amoxicillin: A Broad Spectrum Antibiotic”, International Journal of Pharmacy and Pharmaceutical Sciences, 2011, 3, pp. 30-37.
21. Henry H. Flanner, Modified Release Amoxicillin Products, US patent US2008/0132478A1, 2006, Advancis Pharmaceutical Corporation.2008.
22. medsafe.govt.nz/profs/datasheet/f/Flemoxinsolutab.htm
23. AmnonHoffmana, Haim D. Danenbergb, IfatKatzhendlera, RivkaShuvala, Dalia Gilhar , Michael Friedman, “Pharmacodynamic and pharmacokinetic rationales for the development of an oral controlled-release amoxicillin dosage form”, Journal of Controlled Release, 1998, 54, pp.29–37.
24. HabbanAkhter, NitinSaigal, SanjulaBaboota, Shah Faisal, Javed Ali, “A two pulse drug delivery system for amoxicillin: An attempt to counter the scourge of bacterial resistance against antibiotics”, Acta Pharm., 2011, 61, pp.313–322.
25. emedexpert.com/compare/compare.shtml
26. Indian Pharmacopoeia, The Indian Pharmacopoeia Commission, Ghaziabad, Govt. of India Ministry of Health and Family Welfare, 2007, 2, pp. 722-25.
27. drugbank.ca/drugs/DB01060
28. sitemaker.umich.edu/mc10/amoxicillin
29. Gregory E. Amidon, PhD, “Proposed New USP General Information Chapter, Excipient Performance (1059)”, Pharmacopeial Forum, Nov.–Dec. 2007, 33, 1311-1323.
30. Igor Legen ,Matja Z., MatejaSalobir, Janez Ker C. , “The evaluation of some pharmaceutically acceptable excipients as permeation enhancers for amoxicillin”, International Journal of Pharmaceutics, 2006, pp. 84–89.
31. M.K.Goyal and S.C.Mehta, “Preparation and evaluation of calcium silicate based floating microspheres of amoxicillin”, Journal of Applied Pharmaceutical Science, 2011, pp. 137-141.
32. SualehaRiffat, doctor of philosophy thesis, “bioavailability and disposition kinetics of amoxicillin in normal, febrile, metabolically altered and water deprived rabbits”, university of panjab, 1993.
33. Moxatag information, December 2008. rxlist.com/moxatag-drug.htm - Cached - Similar
34. Raymond C Rowe, Paul J Sheskey and Siân C Owen, Handbook of pharmaceutical excipients, Fifth Edition, Published by the Pharmaceutical Press, USA ,2006.
35. files.shareholder.com/.../MBRK_News_2009_3_16_General.pdf
36. K. Mallikarjuna Rao, K. Gnanaprakash, K.B. Chandra Sekhar, C.MadhusudhanaChety, “Formulation and in-vitro characterization of floating microspheres of amoxycillin trihydrate agaistH.pylori” , Journal of Pharmacy Research, 2011,4, pp.836-840.
37. JayeshParmar, Ph. D., Manish Rane, Ph. D., Viena Dias, M. Pharm., and Ali Rajabi-Siahboomi, Ph. D., “Formulation of Extended Release Multiparticulate Systems using Ethylcellulose”, Pharma Times, April 2010, 42, pp.34-39.
38. Singhal M., Gupta V., Jindal S., Sharma A., Surenderkaswan, NituBishnoi, “Design and Development of Extended Release Oral Solid Dosage Form of Perfenazine”, Journal of NaturaConscientiaMarch 2011, 2, pp.350-362
39. Shiva Kumar Yellanki, Jeet Singh, Jawad Ali Syed, RajkamalBigala, SharadaGoranti, Naveen Kumar nerella, “International Journal of Pharmaceutical Sciences and Drug Research”, 2010, 2, pp.112-114
40. Grimmettet al,Tablet containing a coated core, U S patent 6838094B2, 2001, Smithkline Beecham p.l.c. 2005.
41. Ramarajuet al, Multi-layered modified release formulation comprising amoxicillin and clavulanate, U S patent 2011/0020408 A1, 2007, Ranbaxy Laboratories Limited, 2011.
42. Gregory E. Amidon, PhD, “Proposed New USP General Information Chapter, Excipient Performance (1059)”, Pharmacopeial Forum, Nov.–Dec. 2007, 33, 1311-1323.
43. Igor Legen ,Matja Z., MatejaSalobir, Janez Ker C. , “The evaluation of some pharmaceutically acceptable excipients as permeation enhancers for amoxicillin”, International Journal of Pharmaceutics, 2006, pp. 84–89.
44. M.K.Goyal and S.C.Mehta, “Preparation and evaluation of calcium silicate based floating microspheres of amoxicillin”, Journal of Applied Pharmaceutical Science, 2011, pp. 137-141.
45. Raxit Y. Mehta, Sandip B. Tiwari, Tim Cabelka, Harold Bernthal, Thomas P. Farrell, and Ali Rajabi-Siahboomi, “The Utility of Ultra-High Viscosity Hypromellose in Extended Release Matrix Formulations”, Colorcon, Inc., 2010.
46. KultidaSongsurang, JatupornPakdeebumrung, NarongPraphairaksit, and NongnujMuangsin, “Sustained Release of Amoxicillin from Ethyl Cellulose-Coated Amoxicillin/Chitosan–Cyclodextrin-Based Tablets”, American Association of Pharmaceutical Scientists, March 2011, 12, pp. 35–45.
47. HN Shivakumar, Sarasija Suresh, BG Desai, “Design and evaluation of pH sensitive multi-particulate systems for chronotherapeutic delivery of Diltiazem hydrochloride”, Scientific Publication of the Indian Pharmaceutical Association, 2006, 68, pp. 781-787.
48. MdARahman, J Ali, “Development and in vitro evaluation of enteric coated multiparticulate system for resistant tuberculosis”, Scientific Publication of the Indian Pharmaceutical Association, 2008, 70, pp. 477-481.
49. MuniyandySaravanan, Kalakonda Sri Nataraj, KettavarampalayamSwaminath Ganesh, “The Effect of Tablet Formulation and Hardness on in Vitro Release of Cephalexin from Eudragit L100 Based Extended Release Tablets”, Biol. Pharm. Bull, April 2002, 25, 541—545.
50. United state Pharmacopoeia, USP30 NF-25, The official compendia of standard, 2010, 31, 618, 909.
51. Indian Pharmacopoeia, The Indian Pharmacopoeia Commission, Ghaziabad, Govt. of India Ministry of Health and Family Welfare, 2010, 1, pp. 192-193.
52. accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults_Dissolutions.cfm?PrintAll=1
53. Huey LJ, Shu L., “On the assessment of similarity of drug dissolution profiles- a simulation study”, Drug Information Journal, 1997; 31, pp. 1273-1289.
54. MukeshC.Gohel, Krishnakant G. Sarvaiya, NeelimaR.Mehta, Chirag D. Soni, Vinita U.Vyas, Rikita K. Dave, “Assessment of Similarity Factor Using Different Weighting Approaches”, Dissolution Technologies, November 2005, pp.22-27.
55. Kewal K. Jain, Drug Delivery Systems, humana press USA, 2008, pp 231-232.
56. Suvakanta Dash, PadalaNarasimha Murthy, LilakantaNath, PrasantaChowdhury, “Kinetic modeling on drug release from controlled drug delivery systems”, ActaPoloniaePharmaceutica-Drug Research, 2010, 67, pp. 217-223.
57. Who Guidelines, Guidance for industry, stability testing of new drug substance and products [cited 2011 Apr 12] Available from URL: who.org.
58. David JM. International stability testing. USA: Interpharm Press Inc.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






