
{ DOWNLOAD AS PDF }
 ABOUT AUTHOR
ABOUT AUTHOR
Aman Mittal
Smt. Tarawati Institute of Biomedical & Allied Science
Roorkee, Uttarakhand, India
amanmittal_27@yahoo.com
ABSTRACT
Floating drug delivery systems (FDDS) are the drug delivery systems having a bulk density lower than the gastric content and they remain buoyant in the stomach for a prolonged period of time, with the potential for continuous release of drug. Cinnarizine, a H1-receptor antagonist, used for the treatment for vestibular vertigo disorders and motion sickness was selected as the drug aspirant and Gelucire 43/01 was selected as a lipid carrier in different ratio (1:0.5, 1:1, 1:1.5) along with drug to be formulated as gastro retentive multiparticulate system. The formulation F1 to F9 were prepared and formulation F4 to F9 were evaluated for in-vitro drug release and formulation F5 was selected as optimized formulation that exhibited good floating ability and zero order drug release (93.56 %) at the end of 8 hrs. The in-vitro drug release study of the aged sample show phase conversion of Gelucire. The phase conversion also caused increase in drug release. In conclusion, hydrophobic lipid, Gelucire 43/01 can be considered as an effective carrier for design of a multiple unit floating drug delivery system of cinnarizine.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2429
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 4, Issue 8 Received On: 01/03/2016; Accepted On: 03/04/2016; Published On: 01/08/2016 How to cite this article: Mittal A; Development and In-Vitro Drug Release Profile of Sustained release Floating Granules of Cinnarizine; PharmaTutor; 2016; 4(8); 27-35 |
INTRODUCTION
In general, the gastro retentive time (GRT) of dosage form and in particular large dosage form is longer in the fed state in comparison to the fasting state. Large dosage forms are retropelled from the pyloric-antrum for further digestion and evacuation in the end of the fed state, or are retained until the arrival of the subsequent “house keeper waves”. In such cases, the GRT is a function of the digestive process. Thus, theoretically continuous feeding can prolong GRT of the dosage form for more than 24 hrs[1].
FACTORS AFFECTING GASTRIC RETENTION
· Density of a dosage form affects the gastric emptying rate. A buoyant dosage having a density less than that of the gastric fluid floats. Since, it is away from the pyloric sphincter, the dosage unit in the stomach for a prolonged period.
· The pH of the stomach in fasting state is 1.5 - 2.0 and in fed state 2.0- 6.0. A large volume of water is administered with an oral dosage form raises the pH of the stomach contents to 6.0-9.0. As the stomach does not gets enough time to produce sufficient acid when the liquid empties the stomach, generally basic drug have a better chance of dissolving in the fed state than in a fasting state.
· Size and shape of dosage form also affect gastric emptying time. Tetrahedron and ring shaped devices have a better GRT as compared to other shapes. Dosage form having a diameter of more than 7.5mm2.
· To pass through the pyloric valve into small intestine the particle size should be in the range of 1-2 mm.
· The rate of gastric emptying depends mainly on viscosity, volume and caloric content of meals. Nutritive density of meals helps to determine the gastric emptying time. Increase in acidity and caloric value slows down gastric emptying time.
· Volume of liquid administered affects the gastric emptying time. When the volume is large, the emptying is faster. Fluids taken at body temperature leave the stomach faster than colder or warmer fluids.
· Floating dosage forms are less likely to be expelled from the stomach compared with the non-floating units, which lie in the antrum region and are propelled by the peristaltic waves[3].
· The non-floating units stayed in the lower part of the distal stomach where they were exposed to expulsion through the pylorus as the meal is progressively emptied under the action of the antral peristaltic waves. When their size was larger than the pyloric opening, the sinking forms were retropelled into the antrum and awaited for the powerful contraction.
· More reliable gastric emptying patterns are observed for multi particulate system which are disturbed freely throughout the GIT, further their transport is lesser affected by a transit time of food compared with single unit formulation.
· Residence time can be significantly increased under fed conditions since the MMC is delayed[4].
APPROACHES TO INCREASE GASTRIC RETENTION
Various approaches have been worked out to improve the retention of an oral dosage form in the stomach namely
1. Floating System- Floating drug delivery systems have a bilk density lower than gastric fluids and therefore remain floating in the stomach unflattering the gastric emptying rate for a prolonged period.
2. Swelling And Expanding System- Swellable system include the products that swell after swallowing to an extent that prevents their exit from the stomach through the pylorus.
3. Modified Shaped System-Modified system are non-disintegrating geometric shapes made up of silastic elastomer or extruted from polyethylene blends, which prolonged the GIT depending upon size and shape.
4. Bioadhesive System-In the bioadhesive system, bioadhesive polymers are used that can adhere to the epithelial surface of GIT. Mechanisitically, bioadhesion involves the formation of hydrogen and electrostatic bonding at the mucus polymer interface.
5. High Density System- High density gastroretentive systems include coated pellets that have a density greater than stomach contents (1.004 – 1.010 g/cm3).
6. Delayed Gastric Emptyingf Devices-These include feeding of some indigestablepolymers or fatty acidsalts, which can change the motility of GIT leading to increase in GRT and hence prolonged drug release.
[adsense:468x15:2204050025]
ADVANTAGES ASSOCIATED WITH FDDS
- Slow release of drug at a desired rate from the system.
- Expulsion of the floating system from stomach after complete release of drug
- Reduction in dosing frequency
- Increase in gastric retention time (GRT)
- Increased patient compliance
- Reduction in fluctuations in plasma drug concentration.
- Controlled administration of the therapeutic dose at a desirable delivery rate.
LIMITATIONS of FDDS
- FDDS require sufficiently high levels of fluids in the stomach for drug delivery i.e. 200-500 ml of glass of water is needed for administration.
- Not suitable for the drugs having solubility or stability problems in gastric fluids.
- The drugs which are well absorbed along the entire GIT and undergo extensive first pass metabolism are not desired candidates for FDDS
- The drugs that cause irritation to gastric mucosa are unsuitable for FDDS
MATERIAL & METHODS
Materials: Cinnarizine (Glenmark pharmaceuticals, baddi); Gelucire 43/01, Gelucire 50/13 (Gattefosse, St Priest, Cedex, France); Acetone, Potassium chloride, Hydrochloric acid, Potassium dihydrogen phosphate, Sodium hydroxide pellets, ethanol (Qualigens Fine chemicals, Mumbai); Sodium lauryl sulphate, N-N Dimethyl formamide (Qualigens Fine chemicals Pvt. Ltd., New Delhi) were purchase fro the sources indicated.
METHOD
PREPARATION OF GRANULES OF CINNARIZINE
The granules of cinnarizine were prepared by using the following methods.
1. MELT GRANULATION METHOD- Lipid was melted at 50-60oC and the drug was added, mixed well, and cooled to room temperature. The mass was passed through a 710um (22 mesh) sieve to obtain uniform sized granules.
2. MELT SOLIDIFICATION METHOD- The drug and lipid polymer were melted on a water bath maintained at 100-110oC, stirred it for uniform molten mass, and cooled at 5oC using ice. The mass was passed out through a 710 um (22 mesh) sieve to obtain uniform sized granules. The formulation codes of the granules prepared are listed in below table.
Table 1- formulation design of cinnarizine granules
|
Method of preparation |
Formulation code |
Drug (cinnarizine) |
Gelucire 50/13 |
Gelucire 43/01 |
|
Melt granulation method |
F1 |
1 |
0.5 |
- |
|
F2 |
1 |
1 |
- |
|
|
F3 |
1 |
1.5 |
- |
|
|
F4 |
1 |
- |
0.5 |
|
|
F5 |
1 |
- |
1 |
|
|
F6 |
1 |
- |
1.5 |
|
|
Melt solidification method |
F7 |
1 |
- |
0.5 |
|
F8 |
1 |
- |
1 |
|
|
F9 |
1 |
- |
1.5 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
EVALUATION
DETERMINATION OF IN-VITRO FLOATING ABILITY
20 unit granules were placed in 900ml of simulated gastric fluid (pH 1.2 buffers) and phosphate buffers pH 2.5, 4.5 & 6.5 in a vessel maintained at 37oC and stirred at 100 rpm in USP 24 type II dissolution test apparatus. The percentage of floating granules up to 8 hours was determined and the floating times were measured by visual observation[5].
% of floating ability = Nf / (Nf + Ns ) * 100
Where, Nf and Ns are numbers of the floating and settled granules respectively.
IN-VITRO DRUG RELEASE STUDIES
Dissolution of CN from different formulations were studied in 900 ml pH 1.2 (0.1 N HCl) buffer and phosphate buffers pH 2.50, 4.50 & 6.50 using a USP apparatus 2 (paddle type) dissolution rate test apparatus. Samples equivalent to 50 mg of cinnarizine were used in each test at 100 rpm and temperature 37oC. Samples (5ml) were withdrawn at predetermined time intervals till 8 hours, immediately replaced with fresh dissolution medium and analyzed for CN content at 254 nm after suitable dilution. Percent of CN dissolved at various time intervals was calculated from the regression equation generated from the suitably constructed calibration curve. All the release studies were conducted in triplicates.
IN-VITRO DRUG RELEASE STUDY IN MODIFIED DISSOLUTION MEDIUM
Dissolution of CN from different formulations were studied in 900 ml pH 1.2 (0.1 N HCl) buffer and phosphate buffers pH 2.50, 4.50 & 6.50 containing 0.02% w/v SLS using a USP apparatus 2 (paddle type) dissolution rate apparatus. Samples equivalent to 50 mg of cinnarizine were used in each test at 100 rpm and temperature 37oC. Samples (5ml) were withdrawn at predetermined time intervals till 8 hours, immediately replaced with fresh dissolution medium and analyzed for CN content at 254 nm after suitable dilution. Percent of CN dissolved at various time intervals was calculated calibration curve. All the release studies were conducted in triplicates.
MODEL FITTING
Values obtained from drug release from different formulations in both dissolution conditions i.e. without and with 0.02% w/v SLS in different dissolution mediums (pH 2.5, 4.5 & 6.5) were subjected for model fitting parameters (zero order, first order, Higuchi, Hixson-Crowell and Peppas).
RESULT & DISCUSSION
IN-VITRO DRUG RELEASE STUDIES
The in-vitro drug release profile of the granules prepared by melt granulation (F4-F6) and melt solidification (F7-F9) were compared with that of pure drug. The cumulative percentage release data was shown in Table 2, Table 3, Table 4 & Table 5.
In fasting gastric state, pH (pH 1.2) the release rate of pure cinnarizine showed 99.95% while formulations F4, F7 showed 94.15%, 86.20%, formulations F5, F8 showed 80.68%, 82.02% and formulation F6 and F9 showed 75.59%, 77.10% at the end of 8 hours respectively.
In the fed state, gastric pH ranges from not more than pH 2.0- pH 6.5[6] therefore pH 2.5 and pH 6.5 were selected as extreme fed state gastric pH while pH 4.5 was taken as intermediate fed state gastric pH.
In fed state. At pH 2.5, the pure drug showed 99.01% while formulation F4, F7 showed 75.19%, 76.90% and formulation F5, F8 showed 72.52% and F6, F9 showed 68.52%, 71.12% respectively.
At pH 4.5, the pure drug showed 68.05% while formulation F4, F7 showed 44.20%, 46.52% and formulation F5, F8 showed 42.50%, 44.29% and F6, F9 showed 39.01%, 40.59% respectively.
At pH 6.5, the pure drug showed 64.08% while formulation F4, F7 showed 42.50%, 44.56% and formulation F5, F8 showed 40.56%, 42.54% and F6, F9 showed 36.12%, 38.10% respectively.
The reduction in release rate of pure cinnarizine and formulations F4- F9, when pH lowers was attributed to basic nature (pKa 7.0) of drug that showed higher solubility at lower pH values.
As the Gelucire 43/01 ratio was increased in the formulations F4 to F6 and F7 to F9. The release rate was lowered due to hydrophobic nature of Gelucire 43/01 and there was slight incease in release of formulations (F7-F9) prepared by melt solidification than formulations (F7-F9) prepared by melt granulation technique. The analysis of data was done using PCP disso v2.08 software.
IN-VITRO DRUG RELEASE STUDY IN MODIFIED DISSOLUTION MEDIUM
The in-vitro drug release profiles of formulations F4-F9 were performed in dissolution mediums containg 0.0% w/v sodium lauryl sulphate (SLS), having high HLB value (~40). The release rate of formulations increased in dissolution mediums containing SLS than simple dissolutiomn mediums was attributeed to the lowering of interfacial tension between medium and granule surface. The cummulative percentage release data was shown in table 6, Table 7, Table 8 and Table 9. The analysis of data was done using PCP Disso v2.08 software.
RELEASE KINETICS OF PRELIMINARY OPTIMIZED FORMULATIONS
Regression coefficient, n and k values were obtained for zero order, first order, Higuchi. Hixson Crowell and peppas from the values obtained from % drug release (Table 10 & Table 11). The in-vitro drug release profilewere clarified by the data obtained from model fitting. Model dependent parameters showed zero order model as the best fit model. It was conculded that formulation F5 prepared by melt granulation with Drug: Gelucire 43/01 ratio 1:1 folowed zero order release kinetics as the best fit model.
Table 2: In-vitro release data of preliminary optimized formulations in pH 1.2 buffer
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 1.2(0.1 N HCl) buffer |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
58.47 |
25.94 |
18.45 |
17.10 |
26.86 |
19.12 |
17.95 |
|
3 |
2 |
75.52 |
31.59 |
23.97 |
21.59 |
32.20 |
26.15 |
22.10 |
|
4 |
3 |
84.39 |
39.36 |
30.96 |
27.54 |
40.96 |
32.17 |
28.12 |
|
5 |
4 |
92.32 |
50.54 |
40.87 |
36.20 |
51.22 |
42.54 |
37.52 |
|
6 |
5 |
94.35 |
63.12 |
53.92 |
50.52 |
64.59 |
55.42 |
51.42 |
|
7 |
6 |
96.07 |
73.26 |
66.30 |
59.92 |
74.52 |
67.80 |
62.98 |
|
8 |
7 |
98.49 |
79.21 |
74.83 |
69.52 |
80.54 |
76.33 |
73.10 |
|
9 |
8 |
99.55 |
84.15 |
80.68 |
75.59 |
86.20 |
82.02 |
77.10 |
Table 3:In-vitro release data of preliminary optimized formulations in phosphate buffer pH 2.5.
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 2.5 Phosphate buffer |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
32.47 |
21.52 |
17.59 |
16.59 |
22.19 |
18.52 |
17.00 |
|
3 |
2 |
44.87 |
28.52 |
21.92 |
20.03 |
29.52 |
22.54 |
21.15 |
|
4 |
3 |
58.18 |
35.10 |
28.29 |
25.59 |
36.50 |
29.91 |
26.59 |
|
5 |
4 |
69.20 |
46.12 |
38.20 |
32.52 |
47.59 |
40.05 |
33.60 |
|
6 |
5 |
75.27 |
58.59 |
50.15 |
45.12 |
59.63 |
52.10 |
47.12 |
|
7 |
6 |
81.26 |
65.10 |
60.59 |
55.39 |
66.92 |
62.59 |
57.50 |
|
8 |
7 |
84.24 |
69.52 |
65.52 |
63.20 |
71.10 |
68.50 |
65.59 |
|
9 |
8 |
86.15 |
75.19 |
72.52 |
68.52 |
76.90 |
74.19 |
71.12 |
Table 4:In-vitro release data of preliminary optimized formulations in phosphate buffer pH 4.5.
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 4.5 Phosphate buffer |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
22.47 |
15.59 |
14.92 |
14.50 |
16.19 |
15.10 |
15.05 |
|
3 |
2 |
30.51 |
19.54 |
18.50 |
17.56 |
20.52 |
19.36 |
18.12 |
|
4 |
3 |
35.89 |
23.50 |
22.10 |
21.52 |
24.59 |
32.59 |
22.56 |
|
5 |
4 |
42.92 |
29.50 |
28.52 |
26.56 |
30.29 |
29.59 |
27.50 |
|
6 |
5 |
48.72 |
33.59 |
32.15 |
30.50 |
35.10 |
33.82 |
31.59 |
|
7 |
6 |
55.15 |
37.59 |
35.52 |
34.59 |
39.12 |
36.62 |
35.58 |
|
8 |
7 |
60.76 |
42.10 |
40.12 |
38.10 |
43.59 |
41.19 |
39.52 |
|
9 |
8 |
68.05 |
44.20 |
42.50 |
39.10 |
46.52 |
44.29 |
40.59 |
Table 5:In-vitro release data of preliminary optimized formulations in phosphate buffer pH 6.5.
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 6.5 Phosphate buffer |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
21.38 |
15.19 |
14.75 |
14.19 |
15.50 |
14.92 |
15.03 |
|
3 |
2 |
29.10 |
18.56 |
18.10 |
16.56 |
19.20 |
18.82 |
17.80 |
|
4 |
3 |
35.03 |
22.10 |
21.29 |
20.50 |
22.16 |
22.60 |
21.29 |
|
5 |
4 |
40.74 |
28.15 |
27.59 |
25.50 |
29.10 |
28.80 |
26.56 |
|
6 |
5 |
46.84 |
32.52 |
31.12 |
29.59 |
34.12 |
32.90 |
30.82 |
|
7 |
6 |
54.21 |
36.80 |
34.20 |
33.56 |
37.52 |
35.59 |
34.52 |
|
8 |
7 |
57.99 |
41.15 |
38.56 |
35.10 |
42.50 |
40.52 |
37.50 |
|
9 |
8 |
64.08 |
42.50 |
40.56 |
36.12 |
44.56 |
42.54 |
38.10 |
Table 6:In-vitro release data of preliminary optimized formulations in pH 1.2 buffer containing 0.02% w/v SLS
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 1.2(0.1 N HCl) buffer + 0.02%w/v SLS |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
58.60 |
27.12 |
20.96 |
18.92 |
29.02 |
21.46 |
19.10 |
|
3 |
2 |
76.50 |
33.10 |
27.32 |
22.19 |
34.10 |
27.99 |
23.52 |
|
4 |
3 |
86.36 |
43.12 |
33.67 |
29.56 |
42.02 |
34.91 |
30.15 |
|
5 |
4 |
93.89 |
53.19 |
43.58 |
39.59 |
52.12 |
44.55 |
39.50 |
|
6 |
5 |
99.08 |
66.29 |
55.09 |
53.10 |
65.02 |
56.26 |
53.59 |
|
7 |
6 |
99.76 |
75.20 |
70.31 |
64.95 |
76.59 |
71.65 |
65.19 |
|
8 |
7 |
99.92 |
86.12 |
82.86 |
75.52 |
89.52 |
84.36 |
77.10 |
|
9 |
8 |
99.92 |
97.73 |
93.56 |
85.59 |
98.84 |
95.07 |
86.92 |
Table 7:In-vitro release data of preliminary optimized formulations in pH 2.5 buffer containing 0.02% w/v SLS
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 2.5 Phosphate buffer + 0.02%w/v SLS |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
34.56 |
22.50 |
19.09 |
17.15 |
24.19 |
20.10 |
17.90 |
|
3 |
2 |
46.32 |
29.39 |
25.52 |
21.19 |
32.12 |
24.52 |
23.12 |
|
4 |
3 |
59.28 |
37.59 |
31.10 |
27.02 |
38.59 |
31.19 |
28.19 |
|
5 |
4 |
70.14 |
49.56 |
39.59 |
35.25 |
50.50 |
42.59 |
35.20 |
|
6 |
5 |
78.20 |
62.50 |
52.58 |
48.56 |
63.19 |
54.10 |
49.95 |
|
7 |
6 |
87.52 |
72.12 |
66.52 |
60.50 |
73.59 |
68.19 |
62.59 |
|
8 |
7 |
94.10 |
85.13 |
78.92 |
71.52 |
86.92 |
79.10 |
73.56 |
|
9 |
8 |
99.01 |
94.13 |
89.21 |
80.19 |
96.10 |
90.54 |
81.76 |
Table 8:In-vitro release data of preliminary optimized formulations in phosphate buffer pH 4.5 buffer containing 0.02% w/v SLS
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 4.5 phosphate buffer + 0.02%w/v SLS |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
28.54 |
17.50 |
15.59 |
15.52 |
19.12 |
16.52 |
16.12 |
|
3 |
2 |
39.43 |
24.30 |
20.29 |
18.92 |
26.50 |
21.53 |
19.50 |
|
4 |
3 |
49.52 |
30.50 |
25.56 |
23.50 |
32.52 |
27.12 |
24.59 |
|
5 |
4 |
58.10 |
36.52 |
33.36 |
32.12 |
38.19 |
35.52 |
33.20 |
|
6 |
5 |
67.62 |
44.52 |
42.50 |
40.59 |
46.50 |
44.12 |
42.10 |
|
7 |
6 |
75.29 |
54.52 |
52.26 |
52.92 |
58.10 |
54.59 |
54.10 |
|
8 |
7 |
84.72 |
65.12 |
63.19 |
59.92 |
69.52 |
67.19 |
60.52 |
|
9 |
8 |
89.96 |
76.52 |
72.19 |
66.21 |
78.10 |
74.19 |
67.52 |
Table 9:In-vitro release data of preliminary optimized formulations in phosphate buffer pH 6.5 buffer containing 0.02% w/v SLS
|
S.no. |
Time intervals |
Cumulative %age drug release in pH 6.5 Phosphate buffer + 0.02%w/v SLS |
||||||
|
Pure drug |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
||
|
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
1 |
26.38 |
16.60 |
15.19 |
15.21 |
17.51 |
15.59 |
15.83 |
|
3 |
2 |
37.40 |
23.36 |
20.10 |
17.52 |
25.50 |
20.50 |
18.69 |
|
4 |
3 |
48.01 |
29.50 |
24.92 |
22.10 |
30.51 |
26.52 |
23.62 |
|
5 |
4 |
59.50 |
35.59 |
32.12 |
31.12 |
37.12 |
34.50 |
32.15 |
|
6 |
5 |
64.59 |
43.50 |
41.10 |
39.10 |
44.50 |
42.59 |
40.16 |
|
7 |
6 |
71.52 |
53.36 |
50.50 |
49.56 |
55.12 |
53.10 |
51.19 |
|
8 |
7 |
78.82 |
62.19 |
61.50 |
56.50 |
68.10 |
65.52 |
58.80 |
|
9 |
8 |
83.89 |
75.16 |
70.15 |
65.52 |
78.10 |
71.94 |
66.52 |
Table 10: Model dependent parameters of preliminary optimized formulations (F4-F9) for in-vitro release kinetic studies in pH 1.2 buffer
|
S.No. |
Formulations |
Evaluation parameters |
Zero order |
First order |
Matrix model |
Hixson- crowell model |
Korsmeyer-peppas model |
|
1. |
F4 |
R |
0.9736 |
0.9316 |
0.9759 |
0.9764 |
0.9810 |
|
K |
12.3861 |
6.781 |
11.845 |
11.963 |
13.387 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
2. |
F5 |
R |
0.9911 |
0.9775 |
0.9516 |
0.9896 |
0.9802 |
|
K |
10.2279 |
-0.1695 |
24.1024 |
-0.0470 |
13.083 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
3. |
F6 |
R |
0.9909 |
0.9789 |
0.9459 |
0.9888 |
0.9728 |
|
K |
9.4714 |
-0.1481 |
22.2842 |
-0.420 |
14.2704 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
4. |
F7 |
R |
0.9569 |
0.9904 |
0.9836 |
0.9932 |
0.9799 |
|
K |
11.6243 |
-0.2098 |
27.8258 |
-0.0562 |
23.6267 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
5. |
F8 |
R |
0.9879 |
0.9795 |
0.9581 |
0.9909 |
0.9825 |
|
K |
10.5236 |
-0.1778 |
24.8671 |
-0.0489 |
16.7561 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
6. |
F9 |
R |
0.9894 |
0.9756 |
0.9442 |
0.9863 |
0.9695 |
|
K |
9.8008 |
-0.1574 |
23.0567 |
-0.0442 |
14.7932 |
||
|
n |
- |
- |
- |
- |
2.365 |
Table 11: Model dependent parameters of preliminary optimized formulations (F4-F9) for in-vitro release kinetic studies in pH 1.2 buffer (0.02% w/v SLS)
|
S.No. |
Formulations |
Evaluation parameters |
Zero order |
First order |
Matrix model |
Hixson- crowell model |
Korsmeyer-peppas model |
|
1. |
F4 |
R |
0.9736 |
0.9316 |
0.9759 |
0.9764 |
0.9810 |
|
K |
12.3861 |
-0.2635 |
29.5001 |
-0.0653 |
23.7416 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
2. |
F5 |
R |
0.9926 |
0.9292 |
0.9411 |
0.9665 |
0.9749 |
|
K |
11.2869 |
-0.2190 |
26.5047 |
-0.0566 |
16.9292 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
3. |
F6 |
R |
0.9907 |
0.9515 |
0.9343 |
0.9736 |
0.9648 |
|
K |
10.5134 |
-0.1843 |
24.6485 |
-0.499 |
15.4048 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
4. |
F7 |
R |
0.9726 |
0.9195 |
0.9706 |
0.9693 |
0.9684 |
|
K |
12.5337 |
-0.2792 |
29.8230 |
-0.0677 |
24.6489 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
5. |
F8 |
R |
0.9904 |
0.9227 |
0.9446 |
0.9639 |
0.9731 |
|
K |
11.5134 |
-0.2303 |
27.0850 |
-0.0587 |
18.1417 |
||
|
n |
- |
- |
- |
- |
2.365 |
||
|
6. |
F9 |
R |
0.9923 |
0.9519 |
0.9386 |
0.9759 |
0.9687 |
|
K |
10.5002 |
-0.1837 |
24.6396 |
-0.0498 |
15.6272 |
||
|
n |
- |
- |
- |
- |
2.365 |
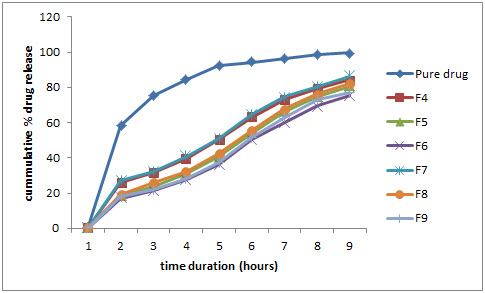
Figure 1: Comparative % drug release profile in pH 1.2 buffer
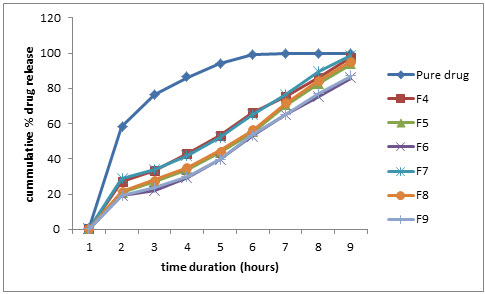
Figure 2: Comparative % drug release profile in pH 1.2 buffer (0.02 % w/v SLS)
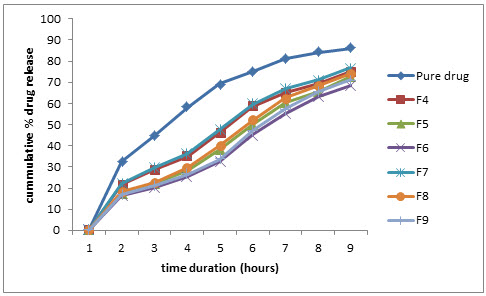
Figure 3: Comparative % drug release profile in pH 2.5 phosphate buffer
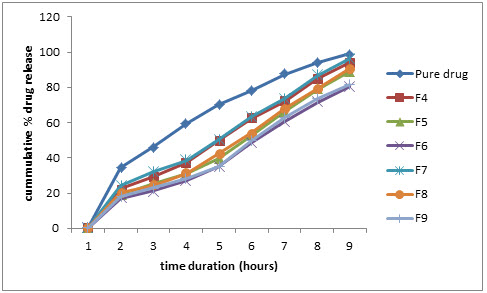
Figure 4: Comparative % drug release profile in pH 2.5 phosphate buffer (0.20% w/v SLS)

Figure 5: Comparative % drug release profile in pH 4.5 phosphate buffer
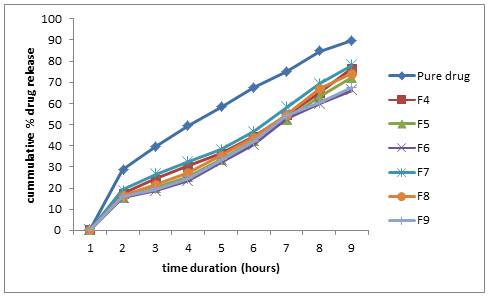
Figure 6: Comparative % drug release profile in pH 4.5 phosphate buffer (0.20% w/v SLS)
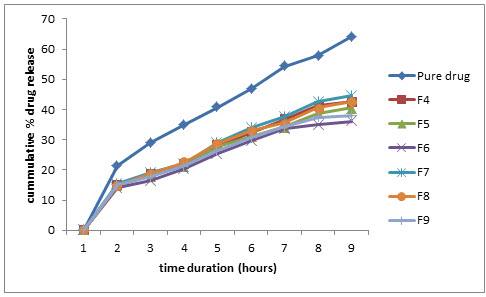
Figure 7: Comparative % drug release profile in pH 6.5 phosphate buffer
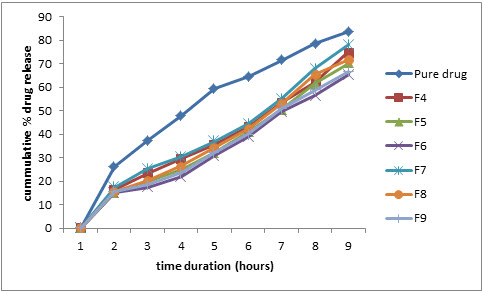
Figure 8: Comparative % drug release profile in pH 6.5 phosphate buffer (0.20% w/v SLS)
CONCLUSION
As the aim of present study was to prepared granules for floating drug delivery system with an objective to control the release rate of cinnarizine. The performance of the formulations was evaluated and the release rate of the drug (cinnarizine) from the granules can be controlled by changing the composition ratio of Gelucire 43/01. formulation F5 (Drug: Gelucire 43/01 ratio 1:1) exhibited good buoyance and drug release (93.56%) with zero order release pattern selected as optimized formulation. The study demonstrated that hydrophobic lipid Gelucire 43/01, can be as an effective carrier for the design of a multi unit floating drug delivery system of cinnarizine.
REFERENCES
1. Read N.W. and Sugden K.; Gastrointestinal dynamics and pharmacology for the optimum design of controlled release oral dosage form; CRT Crit. Rev. Ther. Drug Carrier Syst.; 1987; 4(3); 221-263.
2. Garg S. and Sharma S.; Gastro retentive drug delivery systems, Business Briefing; Pharma Tech., 2003; 160-166.
3. Timmermans J. and Andre J.M.; Factors controlling the buoyancy and gastric retention capabilities of floating matrix capsules: new data for reconsidering the controversy; J. Pharm. Sci.; 1994; 83; 18-24.
4. Mojaverian P., Ferguson R.K. and Vlusses P.H.; Estimation of gastric residence time of the Heidelberg capsules in humans: effect of varying food composition. Gastroenterology; 1985; 89; 392-397.
5. Bhaskar C., Shimpi S., Mahadik K., and Paradkar A.; Preparation and evaluation of floating Risedronate sodium-Gelucire 43/01 formulation; Drug Dev. Ind. Pharm., 2005; 31; 851-860.
6. Arora S., Ali J., Ahuja A., Khar R.K. and Baboota A; Floating Drug Delivery System: A Review; AAPS Pharm. Sci. Tech.; 6(3); E372-E390.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








