

{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
Paras Virani1, 2*, Parul Jain3, Hasumati Raj2, Vineet Jain2
1Research Scholar 2014, Gujrat Technological University, Gujarat
2Department of Quality assurance, Shree Dhanvantary Pharmacy College, Kim, Surat, Gujarat
3Department of Quality assurance, Maliba Pharmacy College, Bardoli, Surat, Gujarat
parasvirani@gmail.com
ABSTRACT
Validation is an act of proving that any procedure, process, equipment, material, activity or system performs as expected under given set of conditions and also give the required accuracy, precision, sensitivity, ruggedness. Validation parameter is used for establishing documented evidence which proves that performance characteristics of the method meet the requirements for the intended analytical applications. The goal of validation is to demonstrate that analytical results produced by the application of a particular method are fit for an intended purpose. In this review article we discussed about the validation and its important parameter.
INTRODUCTION
Method validation is the process of demonstrating that an analytical method is suitable for its intended use, and involves conducting a variety of studies to evaluate method performance under defined conditions. Validation is required for herbal procedure, new process and reaction, new molecules, active ingredients, residues, impurity profiling and component of interest in different matrices. An analytical methodology consists of the techniques, method, procedure and protocol. This methodology includes the required data for a given analytical problem, required sensitivity, required accuracy, required range of analysis and required precision to the analyst.
The International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use[1] has developed a text on the validation of analytical procedures. The United States Food and Drug Administration (USFDA) have proposed guidelines on submitting samples and analytical data for methods validation.[2-5] The United States Pharmacopoeia (USP) has published specific guidelines for method validation for compound evaluation.[5]
The word validation was not mentioned in the current Good Manufacturing Practices (cGMP’s) of 1971, and precision and accuracy were stated as laboratory controls. The need for validation was implied only in the cGMP guideline of March 1979. It was done in two sections: (1) Section 211.165, where the word ‘validation’ was used and (2) section 211.194,[6] in which the proof of suitability,accuracy and reliability was made compulsory for regulatory submissions.
PUBLICATIONS ON VALIDATION (1990 TO PRESENT)
A review on validation of bioanalytical methods was published by Karnes et al. in 1991 which was intended to provide guidance for bioanalytical chemists .[7]One year later, Shah et al. published their report on the conference on "Analytical Methods Validation: Bioavailability, Bioequivalence and Pharmacokinetic Studies" held in Washington in 1990 (Conference Report). During this conference, consensus was reached on which parameters of bioanalytical methods should be evaluated, and some acceptance criteria were established.[8] In the following years, this report was actually used as guidance by bioanalysts And also US FDA Technical Review Guide gives Validation of Chromatographic Methods prepare by Center for Drug Evaluation and Research (CDER) in 1993.[9] That give fully authorized guideline for validation of chromatographic methods.Despite the fact, however, that some principle questions had been answered during this conference, no specific recommendations on practical issues like experimental designs or statistical evaluation had been made. In 1994, Hartmann et al. analyzed the Conference Report performing statistical experiments on the established acceptance criteria for accuracy and precision.[10] Based on their results they questioned the suitability of these criteria for practical application. From 1995 to 1997, application issues like experimental designs and statistical methods for bioanalytical method validation were discussed in a number of publications of Dadgar et al[11], Wieling et al[12], Bressolle et al[13]. and Causon[14]. An excellent review on validation of bioanalytical chromatographic methods has been published by Hartmann et al. in 1998, in which theoretical and practical issues were discussed in detail. Finally, in an update conference of the Washington conference,[15] experiences and progress since the first conference have been discussed. The results were again published by Shah et al. in a report (Conference Report II) .[16] which has also been used as a template for their own guidelines by the U.S. Food and Drug Administration (FDA). Besides, it should be mentioned that some journals like Journal of Chromatography B or Clinical Chemistry have established their own criteria for validation.[17] Two other documents that seem to be important in this context have been developed by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and approved by the regulatory agencies of the European Union, the United States of America and Japan. Despite the fact, that these were focussed on analytical methods for pharmaceutical products rather than bioanalysis, they still contain helpful guidance on some principal questions and definitions in the field of analytical method validation. the first document, approved in 1994[18], concentrated on the theoretical background and definitions, the second, approved in 1996, on methodology and practical issues [19]. both can be downloaded from the ich homepage free of charge (ich.org). validation of analytical methods – strategies & importanceravichandran v, shalini s, sundram k. m and harishrajak in 2010.In few time before gives validation and peer review of u.s. environmental protection agency chemical methods of analysis in 2005,[20] After that validation a critical parameter for quality control of pharmaceuticals tangripranshu, rawatprakashsingh, jakhmolavikash, lakshmayya in 2012,[21] method development and validation- a review article by sudha t, krishanakanth v, nukalapooran in 2012. Guidance for IndustryGuideline for Analytical Procedures and Methods Validation for Drugs and Biologics by FDA and CDER in 2014.[22]
VALIDATION POLICY
1. The company's overall policy, intentions, and approach to validation, including the validation of production processes, cleaning procedures, analytical methods, in-process control test procedures, computerized systems, and persons responsible for design, review, approval and documentation of each validation phase, should be documented.
2. The critical parameters/attributes should normally be identified during the development stage or from historical data, and the ranges necessary for the reproducible operation should be defined. This should include:
- Defining the Medicinal Product/Drug in terms of its critical product attributes;
- Identifying process parameters that could affect the critical quality attributes of the Medicinal Product/Drug;
- Determining the range for each critical process parameter expected to be used during routine manufacturing and process control
3. Validation should extend to those operations determined to be critical to the quality and purity of the Medicinal Product/Drug.
TYPES OF ANALYTICAL PROCEDURES TO BE VALIDATED
the four most common types of analytical procedures:
1. Identification tests
2. Quantitative tests for impurities content
3. Limit tests for the control of impurities
4. Quantitative tests of the active moiety in samples of drug substance or drug product or other selected component in the drug product.
ADVANTAGES OF VALIDATION
The biggest advantage of validation is that it builds a degree of confidence, not only for the developer but also to the user. Although the validation exercise may appear costly and time consuming, it results inexpensive, eliminates frustrating repetitions and leads to better time management in the end. Minor changes in the conditions such as reagent supplier or grade, analytical setup are unavoidable due to obvious reasons but the method validation absorbs the shock of such conditions and pays for more than invested on the process.
IMPORTANCE OF VALIDATION
The most compelling reasons to optimize and validate pharmaceutical productions and supporting processes are quality assurance and cost reduction .the basic principles of quality assurance has as their goal and the production of articles that are fit for their intended use.10 These principles are Quality, safety, and effectiveness must be designed and built in to the product, quality cannot be inspected or tested in the finished products and each step of the manufacturing process must be controlled to maximize the probability that the finished product meets all quality and design specification. The relationship of quality assurance and process validation[23] goes well beyond the responsibility of any quality assurance functions, nevertheless it is fair to say that process validation is a quality assurance tool because it is establishes a quality standard for the specific process.
TYPE OF VALIDATION
· EQUIPMENT VALIDATION
· PROCESS VALIDATION
· CLEANING VALIDATION
· MISCELLANEOUS VALIDATION
· METHOD VALIDATION
EQUIPMENT VALIDATION
Equipment validation is usually carried out by conducting the following activities, individually or combined:
- Design Qualification (DQ): documented verification that the proposed design of the facilities, equipment,or systems is suitable for the intended purpose.
- Installation Qualification (IQ): documented verification that the equipment or systems, as installed or modified, comply with the approved design, the manufacturer’s recommendations and/or user requirements.
- Operational Qualification (OQ): documented verification that the equipment or systems, as installed or modified, perform as intended throughout the anticipated operating ranges.
- Performance Qualification (PQ): documented verification that the equipment and ancillary systems, as connected together, can perform effectively and reproducibly based on the approved process method and specifications.
PROCESS VALIDATION
The number of process runs for validation should depend on the complexity of the process or the magnitude of the process change being considered. For prospective and concurrent validation, three consecutive successful production batches should be used as a guide, but there may be situations where additional process runs are warranted to prove consistency of the process (e.g., complex processes). For retrospective validation, generally data from ten to thirty consecutive batches should be examined to assess process consistency, but fewer batches can be examined if justified.Critical process parameters should be controlled and monitored during process validation studies.Process parameters unrelated to quality, such as variables controlled to minimize energy consumption or equipment use, need not be included in the process validation.
CLEANING VALIDATION
Cleaning procedures should be validated. In general, cleaning validation should be directed to situations or process steps where contamination or carryover of materials poses the greatest risk to bulk product or Medicinal Product/Drug quality.Validation of cleaning procedures should reflect actual equipment usage patterns. If various bulk products or Medicinal Products/Drugs or intermediates are manufactured in the same equipment and the equipment is cleaned by the same process, a representative intermediate or bulk product or Medicinal Product/Drug can be selected for cleaning validation. This selection should be based on the solubility and difficulty of cleaning and the calculation of residue limits based on potency, toxicity, and stability.
METHOD VALIDATION
Method validation guideline has been prepared by the Analytical Methods Technical Committee of the Chemistry Manufacturing Controls Coordinating Committee (CMC CC) of the Center for Drug Evaluation and Research at the Food and Drug Administration.[1]
STEPS IN METHOD VALIDATION
1. Develop a validation protocol.
2. Define purpose and scope of the method
3. Define the performance parameters and acceptance criteria
4. Define validation experiments
5. Verify performance characteristics of equipment
6. Qualify materials, e.g. standards and reagents
7. Perform pre-validation experiments
8. Adjust method parameters
9. Perform full internal (and external) validation experiments
10. Develop SOPs (standard operating procedures) for executing the method in the routine
11. Define criteria for revalidation
12. Define type and frequency of analytical quality control (AQC) checks for the routine
13. Document validation experiments and results in the validation.
PARAMETERS FOR METHOD VALIDATION:
The parameters as defined by the ICH[24] and by other organizations and authors are Specificity, selectivity, precision, intermediateprecision,reproducibility, repeatability, accuracy, stability, recovery, trueness, bias, linearity and calibration model, range, limit of detection, limit of quantitation, robustness and ruggedness.
SELECTIVITY / SPECIFICITY
The terms selectivity and specificity are often used interchangeably. A detailed discussion of this term as defined by different organizations has been made by Vessmann. Even inconsistent with ICH, the term specific generally refers to a method that produces a response for a single analyte only, while the term selective refers to a method which provides responses for a number of chemical entities that may or may not be distinguished from each other. If the response is distinguished from all other responses, the method is said to be selective. Since there are very few methods that respond to only one analyte, the term selectivity is usually more appropriate. The USP monograph 8 defines selectivity of an analytical method as its ability to measure accurately an analyte in the presence of interference, such as synthetic precursors, excipients, enantiomers and known (or likely) degradation products that may be expected to be present in the sample matrix.[12]
Determination OF Selectivity / Specificity:
In the case of qualitative analyses, the ability to select between compounds of closely related structure that are likely to be present should be demonstrated. This should be confirmed by obtaining positive results from samples containing the analyte, coupled with negative results from samples that do not contain the analyte and by confirming that a positive response is not obtained from materials structurally similar to or closely related to the analyte.
Selectivity in liquid chromatography[19] is obtained by choosing optimal columns and setting chromatographicconditions such as mobile phase composition, column temperature and detector wavelength. It is a difficult task in chromatography to ascertain whether the peaks within a sample chromatogram are pure or consist of more than one compound. While in the past chromatographic parameters such as mobile phase composition or the column has been modified.
PRECISIONS
The precision of a method is the extent to which the individual test results of multiple injections of a series of standards agree. The measured standard deviation can be subdivided into three categories: repeatability, intermediate precision and reproducibility.[17] Repeatability is obtained when one operator using one piece of equipment over a relatively short time-span carries out the analysis in one laboratory. At least 5 or 6 determinations of three different matrices at two or three different concentrations should be done and the relative standard deviation calculated.[21]
INTERMEDIATE PRECISION
Intermediate precision is a term that has been defined by ICH-2[26] as the long-term variability of the measurement process and is determined by comparing the results of a method run within a single laboratory over a number of weeks. A method’s intermediate precision may reflect discrepancies in results obtained by different operators, from different instruments, with standards and reagents from different suppliers, with columns from different batches or a combination of these.
Objective of intermediate precision validation is to verify that in the same laboratory the method will provide the same results once the development phase is over.
REPRODUCIBILITY
Reproducibility as defined by ICH-2, 3[24] represents the precision obtained between laboratories. Objective is to verify that the method will provide the same results in different laboratories. Reproducibility means the precision of the procedure when it iscarried out under different conditions-usually in different laboratories-on separate, putatively identical samples taken fromthe same homogenous batch of material. Comparisons of resultsobtained by different analysts, by the use of different equipments, orby carrying out the analysis at different times can also providevaluable information.
REPEATABILITY
Repeatability involves analysis of replicates by the analyst using thesame equipment and method[21] and conducting the precision studyover short period of time while reproducibility involves precisionstudy at different occasions, different laboratories and differentbatch of reagent, different analysts and different equipments.
Determination of repeatability:
It is normally expected that at least six replicates be carried out anda table showing each individual result provided from which themean, standard deviation and co-efficient of variation should becalculated for set of n value. The RSD values are important forshowing degree of variation expected when the analytical procedureis repeated several time in a standard situation. (RSD below 1% forbuilt drugs, RSD below 2% for assays in finished product). The ICHdocuments recommend that repeatability should be assessed using aminimum of nine determinations covering the specified range forthe procedure (i.e. three concentrations and three replicates of eachconcentration or using a minimum of six determinations at 100% ofthe test concentration).
ACCURACY
The accuracy of an analytical method[27] may be defined as thecloseness of the test results obtained by the method to the truevalue. It is the measure of the exactness of the analytical methoddeveloped. The accuracy of an analytical method may be determinedby any of the following ways:
• Analysing a sample of known concentration and comparing themeasured value to the ‘true’ value. However, a wellcharacterized sample (e.g., reference standard) must be used.
• Spiked – placebo (product matrix) recovery method. In thismethod, a known amount of pure active constituent is added toformulation blank [sample that contains all other ingredients except the active(s)], the resulting mixture is assayed, and theresults obtained are compared with the expected result.
• Standard addition method. In this method, a sample is assayed,a known amount of pure active constituent is added, and thesample is again assayed. The difference between the results ofthe two assays is compared with the expected answer.
In both methods (spiked – placebo recovery and standard additionmethod), recovery is defined as the ratio of the observed result tothe expected result expressed as a percentage.
The accuracy of a method may vary across the range of possibleassay values and therefore must be determined at several differentfortification levels. The accuracy should cover at least 3 Concentrations (80, 100 and 120%) in the expected range.
STABILITY
stability of the drug substance or drug product after preparationaccording to the test method should be evaluated according to the testmethod. Most laboratories utilize autosamplers with overnight runs andthe sample will be in solution for hours in the laboratory environmentbefore the test procedure is completed. This is of concern especially fordrugs that can undergo degradation by hydrolysis, photolysis or adhesionto glassware.[25]
RECOVERY
Recovery is expressed as the amount weight of the compound of interestanalyzed as a percentage to the theoretical amount present in themedium.Full recovery[12] should be obtained for the compound(s) of interest. Duringthe sample preparation procedure, the compound of interest is recoveredfrom excipients in the formulation matrix ranging from a simple aqueoussolution to complex cream formulation, and from potential adhesion tocontainer/closure components, e.g., glass vial, metered valve. Ingeneral, a simpler sample preparation procedure will result in a lowervariation of recovery.
BIAS/TRUENESS
Bias refers to the overall magnitude of known systematic (determinate) errors associated with the use of an analytical method. The presence of systematic errors can only be determined by comparison of the average of many results with a reliable, accepted reference value. Method bias may be estimated by measuring materials whose composition is reasonably well known, such as reference materials, by comparing results to those from at least one alternate method or procedure, or by analyzing spiked materials.[22]
The “trueness” of a measurement method is of interest when it is possible to conceive of a true value for the property being measured. Although, for some measurement methods, the true value cannot be known exactly, it may be possible to have an accepted reference value for the property being measured; for example, if suitable reference materials are available, or if the accepted reference value can be established by reference to another measurement method or by preparationof a known sample. The trueness[22] of the measurement method can be investigated by comparing the accepted reference value with the level of the results given by the measurement method. Trueness is normally expressed in terms of bias. Bias can arise, for example, in chemical analysis if the measurement method fails to extract all of an element, or if the presence of one element interferes with the determination of another.
LINEARITY AND CALIBRATION CURVE
The linearity of an analytical method is its ability to elicit test results that are (directly or by means of well-defined mathematical transformations) proportional to the concentration of analytes in samples within a given range. Linearity is determined by a series of three to six injections of five or more standards whose concentrations span 80-120 percent of the expected concentration range. The response should be (directly or by means of a well-defined mathematical calculation) proportional to the concentrations of the analytes. A linear regression equation applied to the results should have an intercept not significantly different from zero. If a significant nonzero intercept is obtained, it should be demonstrated that there is no effect on the accuracy of the method.[28]
The linearity is affected by the various factors like stray light,concentration of sample,wavelength of sample,ultraviolet lamp frequency.[figure 1] represant linearity of sample in case of concentration vs absorbance.
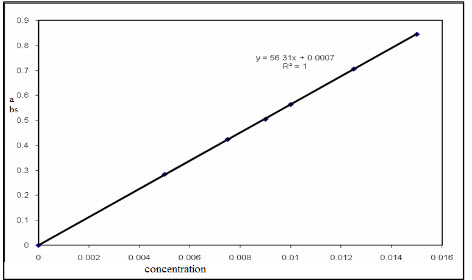
Figure 1 : Linearity of Concentration VsAbsorbance[30]
Figure 2 represent the effect of stray light on the true absorbance of the sample depend on the percentage of stray light interfere with the absorbance of the sample.
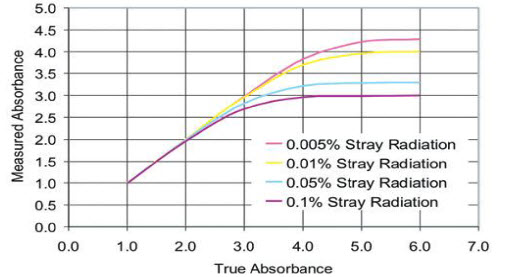
Figure 2. Effect of instrumental stray light on absorbance linearity.[31]
RANGE
The range of an analytical method is the interval between the upper and lower levels (including these levels) that have been demonstrated to be determined with precision, accuracy and linearity using the method as written. The range is normally expressed in the same units as the test results (e.g. percentage, parts per million) obtained by the analytical method.
The range of the method is validated by verifying that the analytical method provides acceptable precision, accuracy and linearity when applied to samples containing analyte at the extremes of the range as well as within the range.[8]
QUANTITATION LIMITS
The term “quantitation range”[17] is used to describe the span of analyte levels, as contained in a sample matrix, for which method performance has been tested, and data quality is deemed acceptable for its intended use. A quantitation range must include either a regulatory or other type of action level. It is the minimum injected amount that gives precise measurements, in chromatography typically requiring peak heights 10 to 20 times higher than baseline noise.
DETECTION LIMIT
The term “detection limit” is used to describe the lowest analyte level that can be confidently identified. There are many specific definitions for this term, and it is used to describe the detection capabilities of detectors, instruments, and analytical methods. The term “detection limit” must be defined, and a description of how it was evaluated during method validation must be provided. Limits derived from mathematical definitions or statistical models must be verified by testing materials containing analyte at the claimed detection level. It is the lowest concentration of analyte in a sample that can be detected but not necessarily quantified. In chromatography the detection limit is the injected amount that results in a peak with a height at least twice or three times as high as the baseline noise level.[29]
ROBUSTNESS
The robustness of an analytical method is a measure of its capacityto remain unaffected by small but deliberate variation in methodparameters and provides an indication of its reliability duringnormal usage. The robustness of a method is evaluated by varyingmethod parameters such as percent organic solvent, pH, ionicstrength, temperature and determine the effect (if any) on theresults of the method. The evaluation of robustness should beconsidered during the development phase and depends on the typeof procedure under study.
If measurements are susceptible to variations in analyticalconditions, the analytical conditions should be suitably controlled ora precautionary statement should be included in the procedure. Oneconsequence of the evaluation of robustness should be that a seriesof system suitability parameters (e.g., resolution test) is establishedto ensure that the validity of the analytical procedure is maintainedwhenever used.Examples of typical variations are stability of analytical solutionsand extraction time[28]
RUGGEDNESS
The ruggedness of an analytical method is the degree ofreproducibility of test results obtained by the analysis of the samesamples under a variety of normal test conditions such as differentlaboratories, different analysts, using operational andenvironmental conditions that may differ but are still within thespecified parameters of the assay. The testing of ruggedness isnormally suggested when the method is to be used in more than onelaboratory. Ruggedness is normally expressed as the lack of theinfluence on the test results of operational and environmentalvariables of the analytical method.
For the determination of ruggedness[25], the degree of reproducibilityof test result is determined as function of the assay variable. Thisreproducibility may be compared to the precision of the assay undernormal condition to obtain a measure of the ruggedness of theanalytical method.
REVALIDATION
A revalidation is necessary whenever a method[21] is changed and thenew parameter is outside the operating range. Operating rangesshould be clearly defined for each method based on experience withsimilar methods, or they should be investigated during methoddevelopments. These ranges should be verified during methodvalidation in robustness studies and should be part of the methodcharacteristics. Availability of such operating ranges makes it easierto decide when a method should be revalidated. If, for example, theoperating range of the column temperature has been specified to bebetween 35 and 40°C, if, for whatever reason, the new operatingparameter has been selected as 42°C, and then the method should berevalidated. Revalidation is also required if the sample matrixchanges and if the instrument type changes.
VALIDATION DOCUMENTATION
A written validation protocol should be established that specifies how validation of a particular process will be conducted. The protocol should be reviewed and approved by the quality unit(s) and other designated units. The validation protocol should specify critical process steps and acceptance criteria as well as the typeof validation to be conducted (e.g. retrospective, prospective, concurrent) and the number of process runs.
A validation report that cross-references the validation protocol should be prepared, summarizing theresults obtained, commenting on any deviations observed, and drawing the appropriate conclusions, including recommending changes to correct deficiencies. Any variations from the validation protocol should be documented with appropriate justification.[26,28]
CONCLUSION
Validation is the first requirement in pharmaceutical company. in addition timely and appropriate validation improves quality assurance ,reduces cost in pharmaceuticals. Analytical methods development plays important roles in the discovery, development and manufacture of pharmaceuticals. Methods should be validated to include consideration of characteristics includedwithin the ICH guidelines on validation of analytical methods. the degree of analytical validation performed should reflect the purpose of the analysis.so in this review that gives few principle, steps, policy of validation and method validation parameter.
REFERANCE
1.Aleem H, Zhao Y, Lord S, McCarthy T and Sharratt P. Pharmaceutical process validation: an overview, Journal of Process Mechanical Engineering,2003; 2(217) :P 141-151.
2.Nash R. A, Wachter A,Marcel Dekkar H. Pharmaceutical Process Validation An International Third Edition. Revised and Expanded, New York, 2003; 129:P760-792.
3.Ramamurthy, M. and Sarvanakumar K. The Eastern Pharmacist.1997;XL 476:P 45-47.
4.UK Orange Guide. Guide to Good Pharmaceutical Manufacturing Practices. 1983:P10.
5.Zutshi. R and Dagar. V, The Eastern Pharmacist, 1991; 34(401),P 37-38.
6. fda.gov/Drugs/.../Guidances/ucm124787.html[online] ,2014.
7.Karnes HT, Shiu G, Shah VP . Validation of bioanalytical methods. journal of Pharmaceutical Research. 1991; 8: P 421-426.
8.Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, Yacobi A, Layloff T, Viswanathan CT, Cook CE, McDowall RD, Pittman KA, Spector S. Analytical methods validation: bioavailability, bioequivalence and pharmacokinetic studies. Conference report Pharma Research. 1992; 9:P 588-592.
9.US FDA Technical Review Guide: Validation of Chromatographic Methods.Center for Drug Evaluation and Research (CDER). Rockville, MD. 1993: P 112- 119.
10.Hartmann C, Smeyers-Verbeke J, Massart DL, McDowall RD. Validation of bioanalytical chromatographic methods. Journal of Pharmaceutical and Biomedical Analysis, 1998; 17:P 193-218.
11.Dadgar D, Burnett PE, Choc MG, Gallicano K, Hooper JW. Application issues in bioanalytical method validation, sample analysis and data reporting, Journal of Pharmaceutical and .Biomedical Analysis.1995; 13: P 89-97.
12.ravichandran v, 1 shalini s,1 sundram k. and harishrajak. validation of analytical methods – importance and strategies. International Journal of Pharmacy and Pharmaceutical Sciences.2010; 5:P112-115.
13.Bressolle F, Bromet PM, Audran M. Validation of liquid chromatographic and gas chromatographic methods Applications to pharmacokinetics. Journal of Chromatography.1996; B 686:P3-10.
14.Causon R. Validation of chromatographic methods in biomedical analysis viewpoint and discussion. Journal of Chromatography. 1997; B 689 :175-180.
15.Hartmann C, Smeyers-Verbeke J,Massart DL, McDowall RD. Validation of bioanalytical chromatographic methods. Journal of Pharmaceutical and Biomedical Analysis. 1998; 17:P 193-218.
16.Shah VP, Midha KK, Findlay JW, Hill HM, Hulse JD, McGilveray IJ, McKay G, Miller KJ, Patnaik RN, Powell ML, Tonelli A, Viswanathan CT, Yacobi A.Bioanalytical method validation--a revisit with a decade of progress.journal of Pharmaresearch. 2000; 17:P 1551-1557.
17.U.S.Department of Health and Human Services and Food and Drug Administration.Guidance for Industry-Bioanalytical Method Validation.2001; fda.gov/cder/guidance/index.htm.
18.International Conference on Harmonization (ICH). Validation of Analytical Methods: Definitions and Terminology. 1994; ICH Q2 A.
19.International Conference on Harmonization (ICH), Validation of Analytical Methods: Methodology. 1996; ICH Q2 B.
20.US EPA. validation and peer review of u.s. environmental protection agency chemical methods of analysis.2005; [online].
21.Tangripranshu, rawatprakashsingh, jakhmolavikash, lakshmayya.validation a critical parameter for quality control of pharmaceuticals. Journal of Drug Delivery & Therapeutics. 2010; 9:P1565-1569.
22. epa.gov/QUALITY/vandv.html ,2014; [online].
23.US FDA AND CDER. Analytical Procedures and Methods Validation for Drugs and Biologics fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm.FEB 2014 ;[ONLINE AND DRAFT COPY].
24.International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use. Validation of analytical procedures: Methodology.Geneva 1996; ICH-Q2B.
25.Wieling J, Hendriks G, Tamminga WJ, Hempenius J, Mensink CK, Oosterhuis B, Jonkman JH. Rational experimental design for bioanalytical methods validation illustration using an assay method for total captopril in plasma. Journal of Chromatography.1996; A 730:P 381-394.
26. ich.org .2014; [online ].
27.US EPA. Guidance for methods development and methods validation for the Resource Conservation and Recovery Act (RCRA) Program. Washington.1995.
28. fda.gov/cder/guidance/index.html .2014 ; [online ].
29.Lindner W, Wainer IW. Requirements for initial assay validation. Journal of Chromatography. 1998; B 707: P 1-2.
30.M. Swamivelmanickam, a.r.gomes, r. Manavalan, d.satyanarayana, p. Gangireddy.Determination and validation of uv spectrophotometric method for estimation of bicalutamide tablet. International journal of chemtech research.2009; 1(4): P 1189-1193.
31.SteveUpstone.Validating UV/Visible Spectrophotometers technical note. perkinelmer publication. 2010: P 4-5.
REFERENCE ID: PHARMATUTOR-ART-2255
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 10 Received On: 22/07/2014; Accepted On: 30/07/2014; Published On: 01/10/2014How to cite this article: P Virani, P Jain, R Hasumati, V Jain; Updated Review: Validation and Method Validation Parameters; PharmaTutor; 2014; 2(10); 27-37 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






