
 About Authors:
About Authors:
Patel Chirag J*1, Pinkesh Patel1, Prof. Satyanand Tyagi2, Tarun Parashar3, Soniya3, Roopesh Sachan3, Gaurav Singh3
1Department of Pharmaceutics, Maharishi Arvind Institute of Pharmacy, Mansarovar, Jaipur, Rajasthan, India-302020.
2President & Founder, Tyagi Pharmacy Association (TPA) & Scientific Writer (Pharmacy), Chattarpur, New Delhi, India-110074.
3Himalayan Institute of Pharmacy and Research, Rajawala, Dehradun, Uttarakhand, India-248007.
*chirag.bangalore@gmail.com, +91-8000501871
ABSTRACT:
Alzheimer's disease is a progressive neurologic disease of the brain leading to the irreversible loss of neurons and the loss of intellectual abilities, including memory and reasoning, which become severe enough to impede social or occupational functioning. Alzheimer's disease is also known as simply Alzheimer’s, and Senile Dementia of the Alzheimer Type (SDAT). During the course of the disease plaques and tangles develop within the structure of the brain. This causes brain cells to die. Patients with Alzheimer's also have a deficiency in the levels of some vital brain chemicals which are involved with the transmission of messages in the brain - neurotransmitters. Alzheimer's disease is the most common form of dementia. The disease gets worse as it develops - it is a progressive disease. There is no current cure for Alzheimer's, although there are ways of slowing down its advance and helping patients with some of the symptoms. Alzheimer's is also a terminal disease - it is incurable and causes death. According the National Institute on Aging, there are estimated to be between 2.4 million and 4.5 million Americans who have Alzheimer's. β-Secretase is an aspartic-acid protease important in the formation of myelin sheaths in peripheral nerve cells. The transmembrane protein contains two active site aspartate residues in its extra cellular protein domain and may function as a dimer.
Beta-secretase 1 (BACE1) also known as beta-site APP cleaving enzyme 1 (beta-site amyloid precursor protein cleaving enzyme 1), memapsin-2 (membrane-associated aspartic protease 2), and aspartyl protease 2 (ASP2) is an enzyme that in humans is encoded by the BACE1 gene.Beta-secretase 2 is an enzyme that in humans is encoded by the BACE2 gene. BACE2 is a close homolog of BACE1, a protease known to be an important enzyme involved in the cellular pathways that some believe lead to Alzheimer's disease. The aim of present article is to provide in depth knowledge about probable role of enzymes BACE1 and BACE2 in the management of Alzheimer's disease (AD). An attempt is also made to focus on general overview of Alzheimer’sdisease.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1576
INTRODUCTION
ALZHEIMER’S DISEASE (AD)
Alzheimer's disease (AD), also known in medical literature as Alzheimer disease, is the most common form of dementia. There is no cure for the disease, which worsens as it progresses, and eventually leads to death. It was first described by German psychiatrist and neuropathologist Alois Alzheimer in 1906 and was named after him [1]. Most often, AD is diagnosed in people over 65 years of age [2], although the less-prevalent early-onset Alzheimer's can occur much earlier. In 2006, there were 26.6 million sufferers worldwide. Alzheimer's is predicted to affect 1 in 85 people globally by 2050 [3]. Although Alzheimer's disease develops differently for every individual, there are many common symptoms. Early symptoms are often mistakenly thought to be 'age-related' concerns, or manifestations of stress [4].
In the early stages, the most common symptom is difficulty in remembering recent events. When AD is suspected, the diagnosis is usually confirmed with tests that evaluate behavior and thinking abilities, often followed by a brain scan if available. As the disease advances, symptoms can include confusion, irritability and aggression, mood swings, trouble with language, and long-term memory loss. As the sufferer declines they often withdraw from family and society (Fig. 1. A & B) [4, 5, 6].
Gradually, bodily functions are lost, ultimately leading to death. Since the disease is different for each individual, predicting how it will affect the person is difficult. AD develops for an unknown and variable amount of time before becoming fully apparent, and it can progress undiagnosed for years. On average, the life expectancy following diagnosis is approximately seven years [7]. Fewer than three percent of individuals live more than fourteen years after diagnosis [8].
The cause and progression of Alzheimer's disease are not well understood. Research indicates that the disease is associated with plaques and tangles in the brain [9]. Current treatments only help with the symptoms of the disease. There are no available treatments that stop or reverse the progression of the disease. As of 2012, more than 1000 clinical trials have been or are being conducted to find ways to treat the disease, but it is unknown if any of the tested treatments will work. Mental stimulation, exercise, and a balanced diet have been suggested as ways to delay cognitive symptoms (though not brain pathology) in healthy older individuals, but there is no conclusive evidence supporting an effect. Because AD cannot be cured and is degenerative, the sufferer relies on others for assistance. The role of the main caregiver is often taken by the spouse or a close relative. Alzheimer's disease is known for placing a great burden on caregivers; the pressures can be wide-ranging, involving social, psychological, physical, and economic elements of the caregiver's life [10-12]. In developed countries, AD is one of the most costly diseases to society [13].
[adsense:468x15:2204050025]
SYMPTOMPS OF ALZHEIMER'S DISEASE [14].
Dementia symptoms include difficulty with many areas of mental function, including:
*Emotional behavior or personality
*Language
*Memory
*Perception
*Thinking and judgment (cognitive skills)
Dementia usually first appears as forgetfulness.
Mild cognitive impairment is the stage between normal forgetfulness due to aging, and the development of AD. People with MCI (Mild cognitive impairment) have mild problems with thinking and memory that do not interfere with everyday activities. They are often aware of the forgetfulness. Not everyone with MCI develops AD.
Symptoms of MCI include:
*Difficulty performing more than one task at a time
*Difficulty solving problems
*Forgetting recent events or conversations
*Taking longer to perform more difficult activities
The early symptoms of AD can include:
*Difficulty performing tasks that take some thought, but used to come easily, such as balancing a checkbook, playing complex games (such as bridge), and learning new information or routines
*Getting lost on familiar routes
*Language problems, such as trouble finding the name of familiar objects
*Losing interest in things previously enjoyed, flat mood
*Misplacing items
*Personality changes and loss of social skills
As the AD becomes worse, symptoms are more obvious and interfere with your ability to take care of yourself. Symptoms can include:
*Change in sleep patterns, often waking up at night
*Delusions, depression, agitation
*Difficulty doing basic tasks, such as preparing meals, choosing proper clothing, and driving
*Difficulty reading or writing
*Forgetting details about current events
*Forgetting events in your own life history, losing awareness of who you are
*Hallucinations, arguments, striking out, and violent behavior
*Poor judgment and loss of ability to recognize danger
*Using the wrong word, mispronouncing words, speaking in confusing sentences
*Withdrawing from social contact People with severe AD can no longer:
*Understand language
*Recognize family members
*Perform basic activities of daily living, such as eating, dressing, and bathing Other symptoms that may occur with AD:
*Incontinence
*Swallowing problems
PATHOPHYSIOLOGY (NEUROPATHOLOGY, BIOCHEMISTRY AND DISEASE MECHANISM) OF ALZHEIMER'S DISEASE (Fig. 2. A - C) [15-17].
Alzheimer's disease is characterized by loss of neurons and synapses in the cerebral cortex and certain sub cortical regions. This loss results in gross atrophy of the affected regions, including degeneration in the temporal lobe and parietal lobe, and parts of the frontal cortex and cingulate gyrus [18].Studies using MRI and PET have documented reductions in the size of specific brain regions in people with AD as they progressed from mild cognitive impairment to Alzheimer's disease, and in comparison with similar images from healthy older adults [19].
Both amyloid plaques and neurofibrillary tangles are clearly visible by microscopy in brains of those afflicted by AD [20]. Plaques are dense, mostly insoluble deposits of beta-amyloid peptide and cellular material outside and around neurons. Tangles (neurofibrillary tangles) are aggregates of the microtubule-associated protein tau which has become hyper phosphorylated and accumulate inside the cells themselves. Although many older individuals develop some plaques and tangles as a consequence of ageing, the brains of people with AD have a greater number of them in specific brain regions such as the temporal lobe [21]. Lewy bodies are not rare in the brains of people with AD [19]. Alzheimer's disease has been identified as a protein misfolding disease (proteopathy), caused by accumulation of abnormally folded amyloid beta and amyloid tau proteins in the brain [22]. Plaques are made up of small peptides, 39–43 amino acids in length, called beta-amyloid (Aβ). Beta-amyloid is a fragment from a larger protein called amyloid precursor protein (APP), a transmembrane protein that penetrates through the neuron's membrane. APP is critical to neuron growth, survival and post-injury repair[23, 24].
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
In Alzheimer's disease, an unknown process causes APP to be divided into smaller fragments by enzymes through proteolysis. One of these fragments gives rise to fibrils of beta-amyloid, which form clumps that deposit outside neurons in dense formations known as senile plaques [25, 26]. AD is also considered a tauopathy due to abnormal aggregation of the tau protein. Every neuron has a cytoskeleton, an internal support structure partly made up of structures called microtubules. These microtubules act like tracks, guiding nutrients and molecules from the body of the cell to the ends of the axon and back.
A protein called tau stabilizes the microtubules when phosphorylated, and is therefore called a microtubule-associated protein. In AD, tau undergoes chemical changes, becoming hyper phosphorylated; it then begins to pair with other threads, creating neurofibrillary tangles and disintegrating the neuron's transport system [27]. Exactly how disturbances of production and aggregation of the beta-amyloid peptide gives rise to the pathology of AD is not known [28]. The amyloid hypothesis traditionally points to the accumulation of beta-amyloid peptides as the central event triggering neuron degeneration. Accumulation of aggregated amyloid fibrils, which are believed to be the toxic form of the protein responsible for disrupting the cell's calcium ion homeostasis, induces programmed cell death (apoptosis) [29]. It is also known that βA selectively builds up in the mitochondria in the cells of Alzheimer's-affected brains, and it also inhibits certain enzyme functions and the utilization of glucose by neurons [30].
Various inflammatory processes and cytokines may also have a role in the pathology of Alzheimer's disease. Inflammation is a general marker of tissue damage in any disease, and may be either secondary to tissue damage in AD or a marker of an immunological response [31]. Alterations in the distribution of different neurotrophic factors and in the expression of their receptors such as the brain derived neurotrophic factor (BDNF) have been described in AD [32, 33].
BETA-SECRETASE 1 (BACE1)ENZYME (Fig. 3. A) [34].
Introduction
Beta-secretase 1 (BACE1) also known as beta-site APP cleaving enzyme 1 (beta-site amyloid precursor protein cleaving enzyme 1), memapsin-2 (membrane-associated aspartic protease 2), and aspartyl protease 2 (ASP2) is an enzyme that in humans is encoded by the BACE1 gene [35]. β-Secretase is an aspartic-acid protease important in the formation of myelin sheaths in peripheral nerve cells [36]. The transmembrane protein contains two active site aspartate residues in its extra cellular protein domain and may function as a dimer.
Role of BACE1in Alzheimer's disease
Generation of the 40 or 42 amino acid-long amyloid-β peptides that aggregate in the brain of Alzheimer's patients requires two sequential cleavages of the amyloid precursor protein (APP).
Extra cellular cleavage of APP by BACE creates a soluble extra cellular fragment and a cell membrane-bound fragment referred to as C99. Cleavage of C99 within its transmembrane domain by γ-secretase releases the intracellular domain of APP and produces amyloid-β. Since alpha-secretase cleaves APP closer to the cell membrane than BACE does, it removes a fragment of the amyloid-β peptide. Initial cleavage of APP by alpha-secretase rather than BACE prevents eventual generation of amyloid-β. Unlike APP and the presenilin proteins important in γ-secretase, no known mutations in the gene encoding BACE cause early-onset, familial Alzheimer's disease, which is a rare form of the disorder. However, levels of this enzyme have been shown to be elevated in the far more common late-onset sporadic Alzheimer's. The physiological purpose of BACE's cleavage of APP and other transmembrane proteins is unknown. BACE2 is a close homolog of BACE1 with no reported APP cleavage in vivo.
However a single residue mutation in APP reduces the ability of BACE-1 to cleave it to produce amyloid-beta and reduces the risk of Alzheimer’s and other cognitive declines [37, 38].
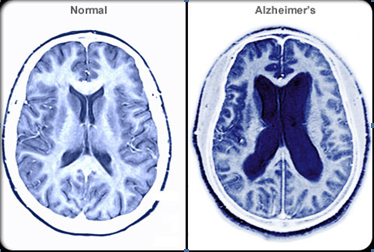
Figure 1. A. Alzheimer’s disease leads to nerve cell death and tissue loss throughout the brain. As the disease progresses, brain tissue shrinks and the ventricles become larger. The damage disrupts communication between brain cells, crippling memory, speech, and comprehension.
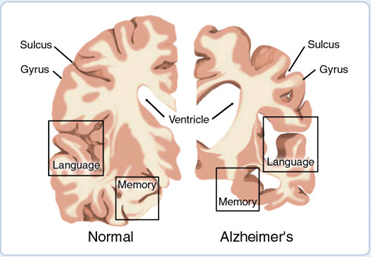
Figure 1. B. Comparison of Normal and Alzheimer’s Brain
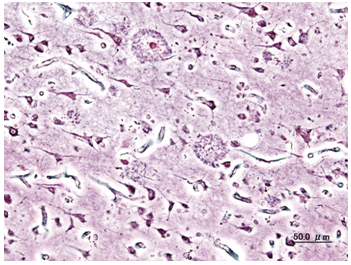
Figure 2. A. Histological image of senile plaques seen in the cerebral cortex in a patient with Alzheimer’s disease of presenile onset. (Silver impregnation)
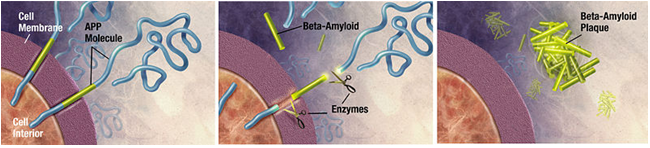
Figure 2. B. Enzymes act on the APP (Amyloid precursor protein) and cut it into fragments of protein, one of which is called beta-amyloid and it’s crucial in the formation of senile plaques in Alzheimer.
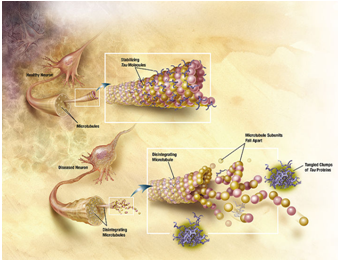
Figure 2. C. Diagram Showing How Microtubules disintegrate with Alzheimer’s disease. In Alzheimer’s disease, changes in tau protein lead to the disintegration of microtubules in brain cells.
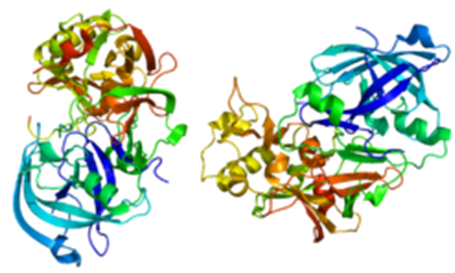
Figure 3. A. Beta-site-cleaving APP enzyme 1 (Structure of BACE1Protein)
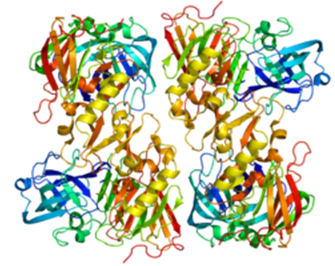
Figure 3. B. Beta-site-cleaving APP enzyme 2 (Structure of BACE2Protein)
BACE Inhibitors
Drugs to block this enzyme (BACE inhibitors) in theory would prevent the build up of beta-amyloid and may help slow or stop Alzheimer’s disease. As of September 2007, several companies are in the early stages of development and testing of this new potential class of treatment [39, 40]. In March 2008 phase I results were reported for CTS-21166. In April 2012 Merck reported phase I results for MK-8931. Side-effects may be caused by blocking the desirable functions of BACE, e.g. in production of myelin.
BETA-SECRETASE 2 (BACE2)ENZYME (Fig. 3. B) [41, 42].
Introduction
Beta-secretase 2 is an enzyme that in humans is encoded by the BACE2 gene [43, 44]. BACE2 is a close homolog of BACE1, a protease known to be an important enzyme involved in the cellular pathways that some believe lead to Alzheimer's disease.
Function
Cerebral deposition of amyloid beta peptide is an early and critical feature of Alzheimer's disease and a frequent complication of Down syndrome. Amyloid beta peptide is generated by proteolytic cleavage of amyloid precursor protein by 2 proteases, one of which is the protein encoded by this gene. This gene localizes to the 'Down critical region' of chromosome 21. The encoded protein, a member of the peptidase A1 protein family, is a type I integral membrane glycoprotein and aspartic protease. Three transcript variants encoding different isoforms have been described for this gene.BACE2 has been shown to interact with GGA1 and GGA2 [45].
RECENT RESEARCH ABOUT BACE2ENZYME [46].
An enzyme that could represent a powerful new tool for combating Alzheimer's disease has been discovered by researchers at Mayo Clinic in Florida. The enzyme — known as BACE2 — destroys beta-amyloid, a toxic protein fragment that litters the brains of patients who have the disease. The findings were published online Sept. 17 in the science journal Molecular Neurodegeneration. The Mayo research team, led by Malcolm A. Leissring, Ph.D., a neuroscientist at Mayo Clinic in Florida, made the discovery by testing hundreds of enzymes for the ability to lower beta-amyloid levels.
BACE2 was found to lower beta-amyloid more effectively than all other enzymes tested. The discovery is interesting because BACE2 is closely related to another enzyme, known as BACE1, involved in producing beta-amyloid. "Despite their close similarity, the two enzymes have completely opposite effects on beta-amyloid — BACE2 giveth, while BACE2 taketh away," Dr. Leissring says. Beta-amyloid is a fragment of a larger protein, known as APP, and is produced by enzymes that cut APP at two places. BACE1 is the enzyme responsible for making the first cut that generates beta-amyloid. The research showed that BACE2 cuts beta-amyloid into smaller pieces, thereby destroying it, instead. Although other enzymes are known to break down beta-amyloid, BACE2 is particularly efficient at this function, the study found. Previous work had shown that BACE2 can also lower beta-amyloid levels by a second mechanism: by cutting APP at a different spot from BACE1. BACE2 cuts in the middle of the beta-amyloid portion, which prevents beta-amyloid production.
"The fact that BACE2 can lower beta-amyloid by two distinct mechanisms makes this enzyme an especially attractive candidate for gene therapy to treat Alzheimer's disease," says first author Samer Abdul-Hay, Ph.D., a neuroscientist at Mayo Clinic in Florida. The discovery suggests that impairments in BACE2 might increase the risk of Alzheimer's disease. This is important because certain drugs in clinical use — for example, antiviral drugs used to treat human immunodeficiency virus (HIV) — work by inhibiting enzymes similar to BACE2. Although BACE2 can lower beta-amyloid by two distinct mechanisms, only the newly discovered mechanism — beta-amyloid destruction — is likely relevant to the disease, the researchers note. This is because the second mechanism, which involves BACE2 cutting APP, does not occur in the brain. The researchers have obtained a grant from the National Institutes of Health to study whether blocking beta-amyloid destruction by BACE2 can increase the risk for Alzheimer's disease in a mouse model of the disease.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
DISCUSSION AND CONCLUSION
Dementia is significant loss of intellectual abilities such as memory capacity, severe enough to interfere with social or occupational functioning. Dementia is reported in as many as 1% of adults 60 years of age. Moreover, it has been estimated that the frequency of dementia doubles every five years after 60 years of age. So, dementia is clearly related to aging.
Alzheimer's disease is the most common form of dementia. Among other causes are medical conditions (thyroid disease, drug toxicity, thiamine deficiency with alcoholism, and others), brain injury, strokes, multiple sclerosis, infection of the brain (such as meningitis and syphilis), HIV infection, hydrocephalus, Pick's disease, and brain tumors. Alzheimer's disease (AD) is a slowly progressive disease of the brain that is characterized by impairment of memory and eventually by disturbances in reasoning, planning, language, and perception. The likelihood of having Alzheimer's disease increases substantially after the age of 70 and may affect around 50% of persons over the age of 85. The main risk factor for Alzheimer's disease is increased age. There are also genetic risk factors and others. There are 10 classic warning signs of Alzheimer's disease: memory loss, difficulty performing familiar tasks, problems with language, disorientation to time and place, poor or decreased judgment, problems with abstract thinking, misplacing things, changes in mood or behaviour, changes in personality, and loss of initiative[47]. Both the enzymes BACE1 and BACE2 are supposed to play prominent role in fighting with Alzheimer's disease. beta-Secretase is the rate limiting enzymatic activity in the production of the amyloid-beta peptide (Abeta) and is thought to be involved in Alzheimer's disease (AD) pathogenesis. Although BACE1 (beta-site APP Cleaving Enzyme 1, EC 3.4.23.46) has received significant attention, the related BACE2 (EC 3.4.23.45) has not. Though BACE2 is also expressed in the brain, its potential role in AD has not been resolved [48].
ACKNOWLEDGEMENT
The corresponding author, Chirag Patel is highly thankful to his Parents & Teachers for their moral support and encouragement. Last but not the least, support of all my friends and the one above all of us, the omnipresent God, for answering my prayers for giving me the strength to plod on despite my constitution wanting to give up and throw in the towel, thank you so much Dear Lord.
REFERENCES
1. Berchtold NC, Cotman CW, Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-Roman period to the 1960’s, Neurobiol. Aging, 1998, 19(3), 173-89.
2. Brookmeyer R, Gray S, Kawas C, Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset, American Journal of Public Health, 1998, 88(9), 1337–42.
3. Brookmeyer R, Johnson E, Ziegler-Graham K, MH Arrighi, Forecasting the global burden of Alzheimer's disease, Alzheimer's and Dementia, 2007, 3(3):186–91.
4. Waldemar G, Recommendations for the diagnosis and management of Alzheimer's disease and other disorders associated with dementia: EFNS guideline, Eur J Neurol, 2007, 14(1), e1–26.
5. Tabert MH, Liu X, Doty RL, Serby M, Zamora D, et al, A 10-item smell identification scale related to risk for Alzheimer's disease, Ann. Neurol, 2005, 58(1), 155-160.
6. ahaf.org/alzheimers/about/understanding/brain-with-alzheimers.html
7. Mölsä PK, Marttila RJ, Rinne UK, Survival and cause of death in Alzheimer's disease and multi-infarct dementia, Acta Neurol Scand, 1986, 74(2),103–7.
8. Mölsä PK, Marttila RJ, Rinne UK, Long-term survival and predictors of mortality in Alzheimer's disease and multi-infarct dementia, ActaNeurol Scand, 1995, 91(3), 159–64.
9. Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J, The importance of neuritic plaques and tangles to the development and evolution of AD, Neurology, 2004, 62(11), 1984-9.
10. Thompson CA, Spilsbury K, Hall J, Birks Y, Barnes C, Adamson J, Systematic review of information and support interventions for caregivers of people with dementia, BMC Geriatr, 2007, 7, 18.
11. Schneider J, Murray J, Banerjee S, Mann A, EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer's disease: I—Factors associated with carer burden, International Journal of Geriatric Psychiatry, 1999, 14(8), 651–661.
12. Murray J, Schneider J, Banerjee S, Mann A. EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer's disease: II—A qualitative analysis of the experience of care giving, International Journal of Geriatric Psychiatry, 1999, 14(8), 662–667.
13. Meek PD, McKeithan K, Schumock GT, Economic considerations in Alzheimer's disease, Pharmacotherapy, 1998, 18(2), 68–73.
14. ncbi.nlm.nih.gov/pubmedhealth/PMH0001767/
15. en.wikipedia.org/wiki/File:Alzheimer_dementia_(3)_presenile_onset.jpg
16. en.wikipedia.org/wiki/File:Amyloid-plaque_formation-big.jpg
17. en.wikipedia.org/wiki/File:Amyloid-plaque_formation-big.jpg
18. Wenk GL, Neuropathologic changes in Alzheimer’s disease, J Clin Psychiatry, 2003, 64(9), 7-10.
19. Desikan RS, Cabral HJ, Hess CP, et al, "Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer's disease", Brain, 2009, 132(8), 2048–57.
20. Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J, The importance of neuritic plaques and tangles to the development and evolution of AD, Neurology, 2004, 62(11), 1984-9.
21. Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH, Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one –year autopsy population from a geriatric hospital, Cereb. Cortex, 1994, 4(2), 138-50.
22. Kotzbauer PT, Trojanowsk JQ, Lee VM, Lewy body pathology in Alzheimer's disease, J Mol Neurosci, 2001, 17(2), 225–32.
23. Hashimoto M, Rockenstein E, Crews L, Masliah E, Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases, Neuromolecular Med, 2003, 4(1-2), 21-36.
24. Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J, Synapse formation and function is modulated by the amyloid precursor protein, J. Neurosci, 2006, 26(27), 7212–21.
25. Turner PR, O'Connor K, Tate WP, Abraham WC, Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory, Prog. Neurobiol, 2003, 70(1), 1–32.
26. Ohnishi S, Tankano K, Amyloid fibrils from the view point of protein folding, Cell. Mol. Life Sci, 2004, 61(5), 511-24.
27. Hernandez F, Avila J, Tauopathies, Cell. Mol. Life Sci, 2007, 64 (17), 2219-33.
28. Van Broeck B, Van Broeckhoven C, Kumar-Singh S, Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches, Neurodegener Dis, 2007, 4(5), 349–65.
29. Yankner BA, Duffy LK, Kirschner DA, Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides, Science, 1990, 250(4978), 279–82.
30. Chen X, Yan SD, Mitochondrial Abeta: a potential cause of metabolic dysfunction in Alzheimer's disease, IUBMB Life, 2006, 58(12), 686–94.
31. Greig NH, New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists, Ann. N. Y. Acad. Sci, 2004, 1035, 290–315.
32. Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S, New insights into brain BDNF function in normal aging and Alzheimer disease, Brain Research Reviews, 2008, 59(1), 201–20.
33. Schindowski K, Belarbi K, Buée L, Neurotrophic factors in Alzheimer's disease: role of axonal transport, Genes, Brain and Behavior, 2008, 7(Suppl 1), 43–56.
34. en.wikipedia.org/wiki/File:Protein_BACE1_PDB_1fkn.png
35. Vassar R, Bennett BD, et al, "Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE", Science, 1999, 286 (5440), 735–41.
36. Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C, "Control of peripheral nerve myelination by the beta-secretase BACE1", Science, 2006, 314 (5799), 664–6.
37. en.wikipedia.org/wiki/Beta-secretase_1#cite_note-APP2012-4
38. Jonsson, et al, A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline, Nature, 2012, 10, 1183.
39. Walker LC, Rosen RF, "Alzheimer therapeutics-what after the cholinesterase inhibitors?”, Age Ageing, 2006, 35 (4), 332-5.
40. Baxter EW, Conway KA, et al, "2-Amino-3, 4-dihydroquinazolines as inhibitors of BACE-1 (beta-site APP cleaving enzyme): Use of structure based design to convert a micromolar hit into a nanomolar lead", J. Med. Chem., 2007, 50(18), 4261-4.
41. en.wikipedia.org/wiki/File:Protein_BACE2_PDB_2ewy.png
42. en.wikipedia.org/wiki/Beta-secretase_2
43. Solans A, Estivill X, De La Luna S, “A new aspartyl protease on 21q22.3, BACE2, is highly similar to Alzheimer's amyloid precursor protein beta-secretase", Cytogenet Cell Genet, 2000, 89 (3-4), 177–84.
44. Hattori M, Fujiyama A, Taylor TD, et al, The DNA sequence of human chromosome 21", Nature, 2000, 405 (6784), 311–9.
45. He Xiangyuan, Chang Wan-Pin, Koelsch Gerald, Tang Jordan, "Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2", FEBS Lett. (Netherlands), 2002, 524 (1-3), 183–7.
46. mayoclinic.org/news2012-jax/7087.html
47. medicinenet.com/alzheimers_disease_causes_stages_and_symptoms/article.htm
48. ncbi.nlm.nih.gov/pubmed/19968762
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








