
{ DOWNLOAD AS PDF }
 ABOUT AUHTOR
ABOUT AUHTOR
SUSMITA DAS*
Department of Pharmaceutical Technology,
Jadavpur University,
West Bengal, India
*sdas53662@gmail.com
ABSTRACT:
Objectives: The aim of the present study was to develop transdermal matrix patches with a fixed ratios of polyvinylpyrrolidone (PVP) and ethylcellulose (EC), containing the drug diclofenac sodium and to perform the physicochemical, in vitro relese pattern evaluation of the prepared patches. The prospective objective was the demonstration that the system provides the delivery of drug at a controlled rate across intact skin to achieve a therapeutically effective drug level for a longer period of time from transdermal patches.
Methods: In this study, matrix-type transdermal patches containing diclofenac sodium were prepared using different ratio of polyvinylpyrrolidone (PVP) and ethylcellulose (EC) by solvent evaporation technique. The drug matrix film of PVP and EC was casted on a polyvinylalcohol backing membrane. All the prepared formulations were subjected to physical studies (moisture content, moisture uptake, and flatness), and in-vitro release kinetics and permeation studies were performed across cadaver skin using a modified diffusion cell.
Results and conclusion: Variations in drug release profiles among the formulations studied were observed. From the formulated patches FA 1, containing PVP/EC in ratio of 1:3 shows sustained release action with 38% release after 24 h and 10 µg of drug permeation accross 1 cm2 of patch area. Hence, it can be reasonably concluded that diclofenac sodium can be formulated into the transdermal matrix type patches to sustain its release characteristics for manufacturing transdermal patches of diclofenac sodium
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2482
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 5, Issue 4 Received On: 20/12/2016; Accepted On: 28/12/2016; Published On: 01/04/2017 How to cite this article: Das S;Preparation and In-Vitro evaluation of Diclofenac Sodium Transdermal Patches; PharmaTutor; 2017; 5(4); 46-54 |
INTRODUCTION
Administration of drugs in the conventional dosage forms usually results in large range fluctuations in plasma drug concentrations leading to undesirable toxicity or poor effectiveness along with limitations such as repetitive dosing and unpredictable absorption, led to the concept of the controlled drug delivery system or therapeutic system.[1] Skin is a well researched and easily approached target which declares its versatility in terms of advantages such as maintenance of constant and prolonged drug level, reduced frequency of dosing, minimization of inter- and intra-patient variability, self administration, and easy termination of medication, leading to patient compliance.[2] Anatomically skin is a multilayered organ comprising three layers from outsides acting as a primary physical immunological barrier.[2,3] The blood flow in different regions are highly variable pertaining to the relative permeability and cellular density which makes the difference in pattern of drug absorption along with several other factors like hydration of skin, contact time and temperature of the area. There are three major routes of penetration across the membrane namely transcorneal, intra follicular and intra appendeageal penetration. On an average, the human skin surface contains 40-70 hair follicles and 200-250 sweat ducts per cm2 and these skin appendages play an important role in the permeation of drug at an early and steady state. [4]
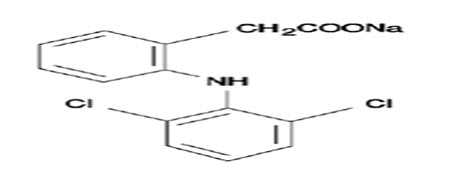
Diclofenac is a widely prescribed nonsteroidal anti-inflammatory drug, widely used in musculoskeletal disorders, arthritis, toothache, dysmenorrhea, etc., for symptomatic relief of pain and inflammation.[5] Sodium salt of diclofenac which is slightly acidic is reportedly used for topical applications and it has a molecular weight of 296.149 which is administered in dose of 25-50 mg oral daily. The limitation pertaining to the oral delivery of the drug undergoes substantial hepatic first-pass metabolism by CYP 450 2C9 and UDP-glucuronosyltransferase and thus only about half of the administered dose reaches systemic circulation.[6]
Transdermal therapeutic systems are defined as medicated dosage forms in form of patch which, when applied to the intact skin, deliver specific dose of drug(s), through the skin, at a controlled rate to the systemic circulation. The drug has a log P value of 4.5 conforming high lipid solubility along with 98% of protein binding. The elimination half-life is 1.2-2 h with clearance of 65% drug unchanged and 35% metabolized in urine. The drug has volume of distribution of 1.4 L. These factors suggests it to be a suitable candidate for incorporation into matrix patches to enhance the delivery.[7]
The first transdermal drug delivery (TDD) system, Transdermal Scop, developed in 1979, used the drug scopolamine for the treatment of motion sickness and over the last two decades more than 35 transdermal patch products have been approved in US. 2004 patch sales in the US were approximately $3.4 billion.[3] Current drugs utilized in TDD systems include nicotine, nitroglycerin and various hormones such as estradiol and testosterone etc and a market analyis is shown in figure 1.
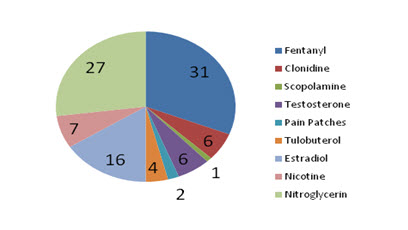
Figure 1: Chart according to the marketability of different patches
Some FDA Approved and widely used marketed Dicofenac patches are FLECTOR® Patch (diclofenac epolamine topical patch)[12] and NuPatch® drug delivery system (India’s first indigenously manufactured patch formulated by Zydus Cadila)[5]
The aim of the present study was to develop different transdermal matrix patches with fixed ratio of polyvinylpyrrolidone (PVP) and ethylcellulose (EC), containing the drug diclofenac sodium and to perform the physicochemical, and in-vitro evaluation of the prepared patches. The purpose was to provide the delivery of drug at a controlled rate across intact skin to achieve a therapeutically effective drug level for a longer period of time from transdermal patches. In this approach, the drug reservoir is formed by homogeneously dispersing of drug solids in a polymeric matrix composed of a suitable blend of PVP K-30 and EC and then molded into medicated disc with a defined surface area and controlled thickness followed by evaluation in-vitro and in-vivo.
MATERIALS AND METHODS:
Diclofenac sodium was obtained as gift sample from Micro Labs Ltd., Hosur, India. Polyvinylpyrrolidone (PVP) (Loba Chemie Pvt Ltd, Mumbai, India), polyvinyl alcohol (PVA) (molecular weight 125,000 Dalton, viscosity of 4% aqueous solution at 20 °C is 23 - 38 cp), tween 80 and ethyl cellulose (EC) (Ethoxy content 47.5–49%, viscosity 14 cps in 5% w/w solution in 80:20 toluene/ethanol at 25°C) (SD Fine-Chem. Ltd., Mumbai, India), dibutyl phthalate (DBP) (Qualigens Fine Chemicals, Mumbai, India) were procured. All the other chemicals used were of analytical grade.
Preparation of backing membrane
The bottom of the mold was wrapped with aluminium foil. Adequately weighted PVA was solubilised into preheated water by slow addition into the water by constant stirring till it forms a homogeneous solution. On the aluminium foil backing membrane was cast by pouring 2% w/v PVA Solution and 4% w/v PVA Solution into glass molds (2.55 cm inner diameter and 2 cm height) followed by drying at 80°C for 24 h. Initially from the above two combinations 4% w/v was screened to achieve appropriate consistency and tensile strength and used for future backing membrane preparation.[8,11,12]
Casting of matrix containing the drug to form transdermal patches:
Matrix-type transdermal patches containing diclofenac sodium were prepared using the 1:2 ratio of PVP and EC by solvent evaporation technique in cylindrical both sides open glass molds (Figure 6). The two polymers were weighed in requisite ratio and they were then dissolved in chloroform. Di-n-Butyphthalate 30% w/w of polymer composition was used as a plasticizer. The drug was added, calculating an amount of 20% w/w of polymer per mold, in the homogeneous dispersion, by sonicating it with an ultra sonicator followed by slow stirring with a mechanical stirrer. 2 mL of the uniform dispersion was cast on the PVA backing membrane cast earlier and dried at 60°C for 24 h. They were kept in desiccators until used.[8,12]
|
Formulation Code |
Rario of PVP/EC |
Total weight of PVP and EC (mg) |
Chloroform (mL) |
di-n-Butylphthalate |
Drug |
|---|---|---|---|---|---|
|
FA 1 |
1:3 |
600 |
10 |
30% w/w of polymers |
20% w/w of polymers |
|
FA 2 |
1:2 |
600 |
10 |
30% w/w of polymers |
20% w/w of polymers |
|
FA 3 |
1:1 |
600 |
10 |
30% w/w of polymers |
20% w/w of polymers |
|
FA 4 |
2:1 |
600 |
10 |
30% w/w of polymers |
20% w/w of polymers |
|
FA 5 |
3:1 |
600 |
10 |
30% w/w of polymers |
20% w/w of polymers |
Table 1: Composition of the prepared batches of transdermal patches
In-vitro evaluation of prepared transdermal patches:
Physicochemical parameters test:
Weight variation test:
Randomly selected twenty patches from each type were directly weighed on a digital balance (Sartorius, GD-103, Goettingen, Germany) and average mass was calculated. Deviations of mass of individual patch from the mean value were determined.[9,10]
Diameter and area of patch:
The diameter (D) of the whole patch (adhesive matrix with the drug(s) plus the backing membrane) was measured at five different randomly selected points of 10 patches using digital calipers (Digmatic Massschieber, CD-6, CSX, Mitutoyo Corp., Japan) in mm scale and average diameter and area (πD2/4) were determined.[11,12].
Thickness of the patches:
The thickness of the backing membrane and the whole patches were measured using digital calipers (Digmatic Masschieber, model CD-6 CS, Mitutoyo, Tokyo). The average thickness of the backing membrane and the whole patch were determined.[11]
Moisture content:
The prepared films were marked, weighed individually and kept in a desiccator containing anhydrous silica at room temperature for 24 h. The mass was taken time to time till it became constant. [11] Percent moisture content was determined as follows:
% Moisture content = X- Y /X × 100 ; where X = initial mass, Y= final mass
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Moisture uptake:
A weighed film kept in a desiccator for 24 h was exposed to 75% relative humidity (saturated solution of sodium chloride) in a desiccator. The film was weighed until constant weight was achieved.[11] Percent moisture uptake was calculated as follows:
% Moisture uptake = X- Y /X × 100 ; where X = initial mass, Y= final mass
Flatness and elongation break:
Longitudinal strips were cut out from the prepared medication film. The flatness was determined at various points by using vernier calipers calculated. The percentage constriction was determined by 0% constriction is equivalent to 100% flatness.[12,13]
Folding endurance:
Evaluation of folding endurance involves determining the folding capacity of the films subjected to frequent extreme condition of folding. Folding endurance was determined by repeatedly folding the film at the same place until it broke. The number of times the film could be folded at the same place without breaking was the folding endurance.[13]
Surface pH study:
The patches were dipped in 1 mL water for 2 min and pH was determined by bringing the electrode in contact by pH meter (Mettler Toledo).
Scanning Electron Microscopy:
The external morphology of the transdermal patches were analyzed using scanning electron microscope (JMS 6100, JEOL, Tokyo, Japan). The samples placed on the stubs were coated finely with gold palladium and examined under microscope.[11]
In-vitro Release Studies:
The dissolution of patches was performed using USP Basket Type Dissolution Apparatus at 32°C and 50 rpm with placement of patches in respective baskets with their drug exposed to phosphate buffer pH 7.4. In 900 mL buffer media, samples are withdrawn at different time intervals and analyzed using a UV spectrophotometer at 276 nm against blank. Cumulative amounts of drug released were plotted against time for different formulations.
In vitro release kinetics:
Data obtained from in-vitro drug release study was plotted in various kinetics models such as zero order, first order, Higuchi, Korsmeyer-Peppas and Hixson-Crowell kinetic models and coefficient of regression and rate constant/release rate exponents for zero order (K0), first order (K1), Korsmeyer-Peppas model and Hixson-Crowell model (KHC) were determined.[11]
In- vitro Permeation Studies:
As a predictive of in-vivo performance of drug, in - vitro permeation studies for the prepared transdermal patches were performed in a modified Keshary-Chien cell using human cadaver skin. The skin was stored was immersed in 40 ml phosphate buffer saline solution, pH 7.4 for 2hr. A section of skin was cut, measured, and placed on the dermal side of the skin in the donor compartment facing the drug matrix side of the patch to the skin and backing membrane upward. The holder containing the skin and formulation was then placed on the receiver compartment of the modified diffusion cell, containing phosphate buffer pH 7.4. This whole assembly was kept on a magnetic stirrer and solution in the receiver compartment was constantly and continuously stirred during the whole experiment using magnetic bead. The samples were withdrawn (1 mL each time) at different time intervals and an equal amount of phosphate buffer, pH 7.4, was replaced each time. Absorbances of the samples were read spectrophotometrically at 276 nm taking phosphate buffer solution, pH 7.4, as blank. The amount of drug permeated per square cm at each time interval was calculated and plotted against time.[12]
RESULTS AND DISCUSSION
Average diameter, area and thickness of the patches and backing membrane
The mean diameter, area and thickness of both the patch and caking membrane were calculated. The result is tabulated in table 2. Thin patches were developed with average thickness varied from 0.978 ± 0.04 to 1.150 ± 0.03 mm. The patches were circular with almost uniform area ranging from 4.789 ± 0.156 to 5.144 ± 0.323 cm2.
|
Formulation Code |
Mean diameter of patch (cm) ± SD (n=10) |
Mean area of patch (cm2) ± SD (n=10) |
Mean thickness of patch (mm) (adhesive matrix + backing membrane) ± SD (n=10) |
Mean thickness of backing membrane (mm) ± SD (n=10) |
|---|---|---|---|---|
|
FA 1 |
2.54 ± 0.051 |
5.071 ± 0.206 |
1.150 ± 0.03 |
0.182 ± 0.04 |
|
FA 2 |
2.47 ± 0.031 |
4.789 ± 0.156 |
1.033 ± 0.01 |
0.187 ± 0.07 |
|
FA 3 |
2.49 ± 0.012 |
4.865 ± 0.184 |
0.978 ± 0.04 |
0.171 ± 0.08 |
|
FA 4 |
2.51 ± 0.039 |
4.950 ± 0.121 |
1.022 ± 0.02 |
0.189 ± 0.05 |
|
FA 5 |
2.56 ± 0.047 |
5.144 ± 0.323 |
0.986 ± 0.03 |
0.177 ± 0.02 |
Table 2: Mean diameter, area, thickness of the patch and the backing membrane of the prepared batches
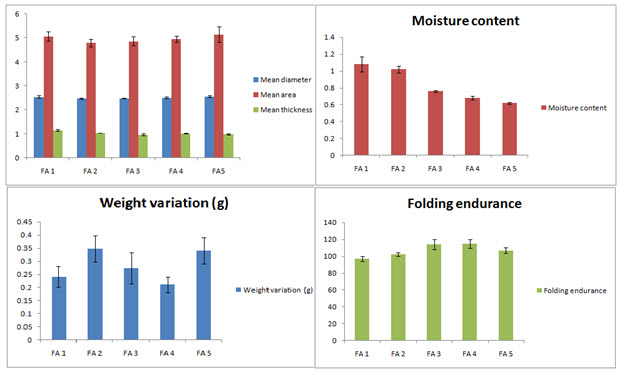
Average weight variation, moisture content of the formulated patches
The formulated patches were thin and almost uniform with low weight variations ranging from 0.211 ± 0.03 g to 0.348 ± 0.03 g. The average moisture content of the patches ranges from 1.08 ± 0.09 % w/w to 0.62 ± 0.01 % w/w. This small amount of moisture present in the patches prevents them from drying, being brittle and unnecessary weight gain
|
Formulation Code |
Mean Weight variation (g) |
Moisture content (% w/w) |
Flatness (%) |
Folding endurance |
|
FA 1 |
0.241 ± 0.04 |
1.08 ± 0.09 |
100 |
97 ± 3.16 |
|
FA 2 |
0.348 ± 0.05 |
1.02 ± 0.04 |
100 |
102 ± 2.11 |
|
FA 3 |
0.274 ± 0.06 |
0.76 ± 0.01 |
100 |
114 ± 6.21 |
|
FA4 |
0.211 ± 0.03 |
0.68 ± 0.02 |
100 |
115 ± 5.12 |
|
FA 5 |
0.341 ± 0.05 |
0.62 ± 0.01 |
100 |
107 ± 3.19 |
Table 3: Mean weight variation, moisture content, flatness and folding endurance of prepared batches.
Folding endurance and flatness of the patches
In order to represent that patches will maintain their integrity for sufficient time after application to the skin folding endurance test was conducted which shows values ranging from 97 ± 5.21 to 114 ± 3.21. Again the patches showed no differences in strip length before and after cuts ensuring 100% flatness and no sign of constriction with time. The representative data are shown in figure 2.
Surface pH The surface pH was ranged from 6.51 to 7.43 which is within the limit of sustainable skin pH and hence no chance of irritation to the site of administration is ensured.
Scanning electron microscopy Scanning electron micrograph of the formulated patches shows homogeneous and uniform distribution of drugs within the matrix ensuring better activity and drug release. The SEM picture is shown in figure 3.

Figure 3: Scanning electron micrograph of prepared transdermal patch (FA 1)
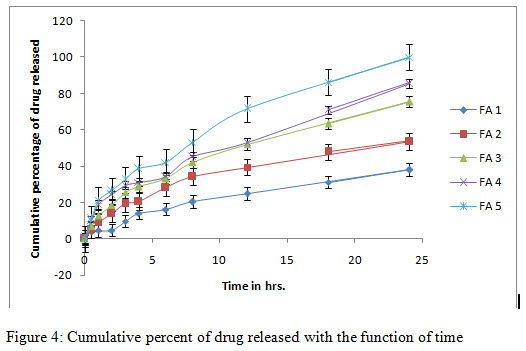
In-vitro release study and release kinetics:
In-vitro release studies shows that the drug is released for a period of 24 h at different rates depending on the composition of matrix, blend of PVP and EC. Release was performed in phosphate buffer pH 7.4 to simulate the condition of the skin. Additionally it has been demonstrated in the release data shown in figure 4 that formulation FA 3 to FA 5 followed release in a rectilinear fashion, within 8 h, for FA 4, almost 40% drug was released due to matrix erosion. Maximum percent of drug released 98% was shown in case of FA 5. They shows zero order release profile as well as first order profile after some period of time. In case of formulation FA 1 and FA 2 very lower amount were released till 24 h showing a sustained release profile throughout the study. In these cases, for FA 2, almost 53% of drug released till the end of study and for FA 1, it could only reach 38% within the time. In regard to the release kinetics, it can be concluded that due to the presence of hydrophobic polymer, matrix erosion followed a rate dependant manner in case of FA 1 and 2, releasing only a lower percent of drug through matrix erosion. On the contrary, due to a higher percent of hydrophilic polymer, more drug was released with time. In figure 5 and 6, various kinetic models, based on the release data are shown. Various rate constants determined for model fitting are tabulated in table 4
|
Kinetic model |
FA 1 |
FA 2 |
FA 3 |
FA 4 |
FA 5 |
|---|---|---|---|---|---|
|
Zero order |
R2= 0.947 K0= 1.529 |
R2= 0.900 K0= 2.135 |
R2= 0.934 K0= 2.933 |
R2= 0.945 K0= 03.166 |
R2= 0.939 K0= 3.865 |
|
First order |
R2= 0.972 K1= -0.008 |
R2= 0.954 K1= -0.013 |
R2= 0.991 K1= -0.023 |
R2= 0.971 K1= -0.030 |
R2= 0.984 K1= -0.044 |
|
Korsmeyer-Peppas |
R2= 0.953 n= 0.715 |
R2= 0.989 n= 0.566 |
R2= 0.995 n= 0.558 |
R2= 0.972 n= 0.470 |
R2= 0.985 n= 0.507 |
|
Higuchi |
R2= 0.980 KH= 8.059 |
R2= 0.992 KH= 11.61 |
R2= 0.996 KH= 15.69 |
R2= 0.986 KH= 16.76 |
R2= 0.992 KH= 20.59 |
|
Hixson-Crowell |
R2= 0.965 KHC= -0.027 |
R2= 0.938 KHC= -0.042 |
R2= 0.981 KHC= -0.067 |
R2= 0.981 KHC= -0.082 |
R2= 0.986 KHC= -0.116 |
Table 4: Various release rate constants determined for all formulation batches on the basis of release data
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
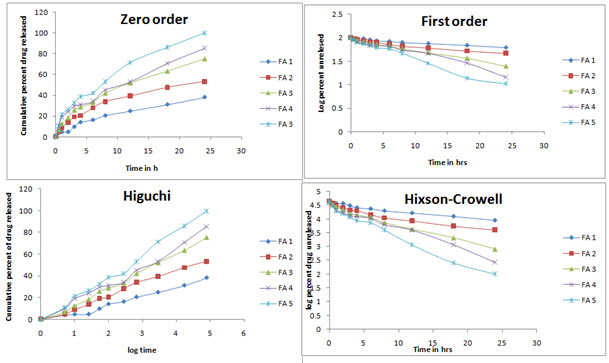
Figure 5: Release kinetics model of the prepared batches in different kinetics profiles
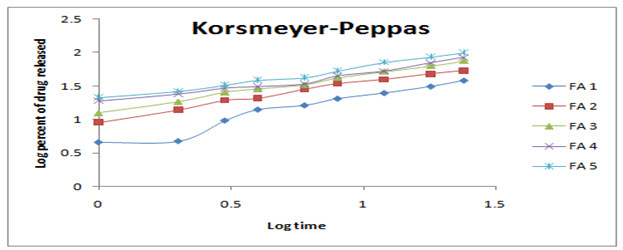
Figure 6: Korsmeyer-Peppas release kinetics model for prepared transdermal patches
In-vitro skin permeation study
In order to show a predictive tool for in vivo performances of the patches, the study was done for different formulations using human cadaver skin using phosphate buffer pH 7.4. The cumulative amount of drug permeated across skin for FA 1 was only 2.48 µg through 1 cm2 area of the patch in first 8h followed by a gradual increase in amount of permeation. For formulation FA 4 to FA 5, drugs were permeated at a faster rate, may be due to rapid matrix erosion and more concentration of drug released triggers faster permeation. The results are in accordance with the in-vitro release data shown in figure 7.

Figure 7: Graph of cumulative amount of drug permeated per unit area with respect to time
CONCLUSION
Diclofenac sodium is a widely used non steroidal anti inflammatory drug which must be substantially reached to the site of action overcoming various absorptional barriers and enzymatic degradation in vivo. In accordance to the objective of the study, the prepared diclofenac sodium transdermal patches were found to release the drugs in a controlled manner in vitro and permeate through the skin to produce local therapeutic actions like pain relief. They were found to posses desired shape with easy administration and removal. Upon the experimental oral formulations, FA 1 showed sustained release activity in terms of both release and permeation showing that a long term pain relief may be achieved. Further study may be necessary in order to produce in vivo data and correlate it with in vitro.
REFERENCES
1.Andrew GP, Lavarty TP, Jones DS. Mucoadhesive polymeric platforms for controlled drug delivery. Eur J Pharm Biopharm. 2009; 71(3):25-29.
2.Prausnitz MR, Langer R. Transdermal drug delivery. Nat Biotechnol. 2008; 26(11):1261-1268.
3.Gaiwak AK. Transdermal drug delivery system: formulation aspects and evaluation Com. J. Pharm. Sci. 2013; 1(1):1-10.
4.Dhiman S, Singh TG, Rehni AK, Transdermal patches: a recent approach to new drug delivery system, Int J Pharm Sci. 2011; 3(5):26-34.
5.Diclofenac transdermal patches: Medline Plus; https://medlineplus.gov/drug info/meds/a611001.html. Accessed on 6th December, 2016.
6.Bucci R, Magri AD, Magri AL. Determination of diclofenac salts in pharmaceutical formulations. Fresnius J Anal Chem. 1998; 362(7):2577-2582.
7.Ozgurey IS, Karasulu HY, Kantarci G. Transdermal delivery of diclofenac sodium through rat skin from various formulations. AAPS Pharm Sci Tech. 2006; 7(4):E1-E7.
8.Mukherjee B, Das S, Patra B, Layek B. Nefopam containing transdermal matrix patches based on pressure sensitive adhesive polymers. Pharm. Technol. 2006; 30(3):146-163.
9. Keith AD. Polymer matrix consideration for transdermal devices. Drug Deliv Ind Pharm. 1983; 9(4):605-621.
10.Sahu RK, Jain A, Nayak S. Development and evaluation of transdermal patches of colchicine. Der. Pharm. Lettre. 2012; 4(1):330-343.
11.Manasadeepa R, Paul P, Mukherjee B. Pressure sensitive mucoadhesive polymer based dental patches to treat periodontal disease: an in vitro study. Drug Deliv. 2013; 20(6):258-267.
12.Arora P, Mukherjee B. Design, development, physicochemical and in vitro and in vivo evaluation of transdermal patches containing diclofenac diethylammonium salt. J Pharm Sci. 2002; 91(9): 2076-2089.
13.Damodharan N, Roy G, Ghosh S, Mukherjee B. Skin permeation of rosiglitazone from transdermal matrix patches. Pharm Technol. 2010; 34(3):56-72
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








