
ABOUT AUTHORS
Md. Nazmul Islam Prottoy1*, Md. Asad Ullah1, Bishajit Sarkar1, Sohana Hossain1, Aisha Siddiqua Boby1, Yusha Araf2
1Dept. of Biotechnology and Genetic Engineering, Jahangirnagar University, Dhaka, Bangladesh
2Dept. of Genetic Engineering and Biotechnology, Shahjalal University of Science and Technology, Sylhet, Bangladesh
ABSTRACT
Diabetes Mellitus (DM) is a metabolic disorder which affects the people of almost all ethnic groups around the world severely. The effect of this disease involves lifelong suffering and has no permanent cure till now. Although different medications are available in the market but they are not accessible to every person due to their high cost, requirement of frequent administration and inability to alleviate diabetes permanently. Plant derived compounds are being commonly used by many people, specifically by those in rural areas of many countries as ayurvedic source of antidiabetic agents and these are more preferable to everyone due to their less toxicity and side effects. These compounds work by variety of mechanisms which involve different interactions between effective compounds and target proteins in the metabolic pathway. Molecular docking study helps in determining the interaction between specific ligands and receptors to specify the best lead that fits the target. This study has been designed to investigate the interactions with the aid of computational simulation tool between medicinal plant derived antidiabetic agents (Aegeline, Gallic Acid, Mangiferin and Quercetin) and a glucose metabolism regulatory target enzyme involved in type II diabetes, Glycogen Synthase Kinase-3 Beta (GSK3B) to assist potential antidiabetic drug search from natural source.
ADME/T test assists in determining various physicochemical and pharmacological properties of lead molecules like their extent of adsorption inside the cell, extent of metabolism, solubility, blood brain barrier permeability, mutagenicity, carcinogenicity etc. which are the major prerequisites before marketing a drug. Quercetin performed well in overall experiment suggesting the best finding of the experiment.
However, further in vitro/in vivo study is required to find out the best remedy of diabetes.
Reference Id: PHARMATUTOR-ART-2680
INTRODUCTION
Diabetes Mellitus, Its Current Status and Treatment
Diabetes is a metabolic disease characterized by hyperglycemia resulting from defects in pancreatic insulin secretion, insulin action, or combination of both. The chronic diabetes is the major cause of dysfunction of organs like eyes, kidneys, nerves, heart, and blood vessels (ADA, 2014). The total number diabetic patient has increased drastically from 180 million in 1980 to 422 million in 2014. The prevalence of patient has increased doubly in past three decades from 4.7% to 8.5% globally. Recent studies have indicated that the occurrence of the disease is now approximately similar in both low income and high income countries (Roglic, 2016). There are different treatments available like- phototherapy, enzyme inhibition, antihypertensive therapy, recombinant insulin administration etc. depending on the type of complications involved in diabetes (Clark and Lee, 1995). Many medicinal plants have been reported to have antidiabetic activity in laboratory experiments (Tapsell et al. 2006; Shukia et al., 2002). Bangladesh is an agricultural country and many plant species are available here with a good portion among them is effective in healing diabetes. Plant species like Aegle marmelos, Terminalia bellirica, Psidium guajava and Mangifera indica are widely used as ayurvedic regimen by the people of rural areas in Bangladesh(Hasan and Sultana, 2018). Aegeline from Aegle marmelos (Narender et al.; 2007), Gallic acid from Terminalia bellirica (Sabu and Kuttan, 2009), Quercetin from Psidium guajava (Cheng et al., 2009) and Mangiferin from Mangifera indica (Apontes et al., 2014) have been shown to be effective in alleviating diabetes in earlier in vitro study.

Figure 1: Chemical structures of ligand molecules. (1) Aegeline; (2) Gallic acid; (3) Mangiferin and (4) Quercetin.

Figure 2: Structure of GSK3B ( PDB ID 1O9U) in its native ligand bound form. The ligand is represented in stick style (yellow) and the receptor is represented as ribbon style.
Role of GSK3B in Glucose Metabolism and Diabetes Development
Glycogen Synthase Kinase-3 Beta (GSK3B) is a multifunctional enzyme (47 kDA) which plays variety of significant roles in cellular signaling. It is usually a serine/threonine kinase which catalyzes the phosphorylation of many cellular mediators and enzymes, and itself becomes activated by tyrosine phosphorylation and inactivated by serine phosphoryltion respectively. (Grimes and Jope; 2001). The phosphorylation carried out by activated GSK3B (Tyr phosphorylated state) is inhibitory for most of its target proteins (Martinez et al.; 2002). GSK3B remains activated when the body is in resting state and phosphorylates and inactivates one of its targets- Glycogen Synthase (GS)- an enzyme that synthesizes glycogen from blood glucose and keeps the glucose concentration lower inside bloodstream. But GSK3B becomes rapidly inactivated when the insulin level increases inside bloodstream and it loses its ability to phosphorylate GS leaving it to synthesize glycogen. The insulin dependent inactivation of GSK3B requires the activation of phosphatidylinositol 3-kinase and protein kinase B (PKB/Akt) (Cross et al., 1995; Sutherland et al., 1993). Dysregulation of GSK3B gives rise to type 2 diabetes which is characterized by the impairment in the insulin dependent glycogen synthesis. In such cases, GSK3B requires to be inhibited by exogeneous inhibitors to alleviate type 2 diabetes (Patel el al., 2008).
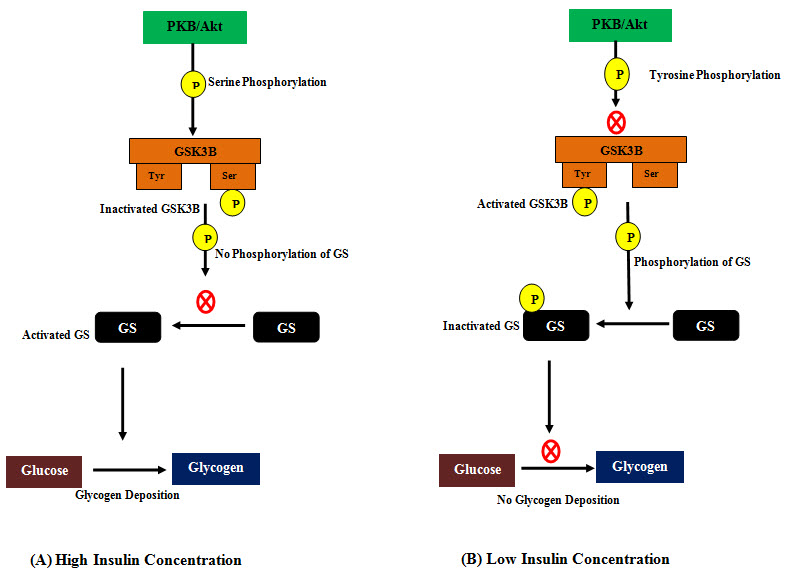
Figure 3: Role of Glycogen Synthase Kinase-3 Beta (GSK3B) in glycogen synthesis.
(A) At higher insulin concentration, activation of phosphatidylinositol 3-kinase and protein kinase B (PKB/Akt) phosphoryaltes GSK3B at serine residue and inactivates it. Inactivated GSK3B can’t phosphporylate and inactivate Glycogen Synthase (GS) and activated GS in turn keeps glycogen synthesis on. (B) At low insulin concentration, inactivated phosphatidylinositol 3-kinase and protein kinase B (PKB/Akt) leaves the GSK3B phosphorylated at tyrosine residue which is its active state. In this state it phosphoryalates and inactivates GS and inactivated GS can’t synthesize glycogen anymore.
In Silico Docking and ADME/T
Molecular docking is an integral tool in structural biology and computer-assisted drug design which helps primarily in determining ligand-protein docking. The aim of molecular docking is to find out the best possible ligand which possesses the predominant binding mode with the 3D structure of a target protein (Morris and Lim-Wilby, 2008). The method works on the basis of specific algorithm which assigns binding energy for each of the ligand molecule from a large library and allows ranking of ligand molecules on the basis of binding energy which reflects its binding affinity. The lowest binding energy determines the most favorable binding (Ferreira et al., 2015).
In silico ADME/T test aids in determining the adsorption, distribution, metabolism, excretion and toxicology of the drug candidate inside the biological system. In addition to those physicochemical properties, ADME/T also focuses on some pharmacological properties like- blood brain barrier permeability, abdominal absorption, carcinogenicity, Cytochrome inhibitory property etc. This process mimics in-vivo condition and assists the subsequent laboratory experiments (Wang and Skolnik, 2009; Dearden, 2007).
This study has been designed to perform molecular docking and ADME/T of four isolated compounds (Arjunic acid, Gallic acid, Quercetin and Mangiferin) from medicinal plants of Bangladesh to identify one that fits well with GSK3B as potent inhibitor and also exhibits sound ADME/T properties.
MATERIALS AND METHODS
Ligand preparation, Grid generation and Glide docking were carried out using Maestro Schrödinger Suite 2018 and the chemical structures of ligands were refined using ChemSketch.
Protein Preparation
Three dimentional structure of GSK3B (PDB id: 1O9U) was downloaded in PDB format from protein data bank (www.rcsb.org). The protein was then processed and refined utilizing the Protein Preparation Wizard in Maestro Schrödinger v11.8. Bond orders were assigned, hydrogens were added to heavy atoms. All the waters were deleted from the molecules and selenomethionines were converted to methionines. Finally, the structure was optimized and then minimized using built in default force field OPLS_2005. Minimization was performed setting the greatest substantial particle RMSD (root-mean-square-deviation) to 30 Å and any outstanding water under 3H-bonds to non water was again erased during the minimization step.
Ligand Preparation
The 3D conformations of Aegeline (PubChem CID: 15558419), Gallic acid (PubChem CID: 370), Mangiferin (PubChem CID: 5281647) and Quercetin (PubChem CID: 5280343) were downloaded from PubChem (www.pubchem.ncbi.nlm.nih.gov). These structures were then processed prepared using the LigPrep of Maestro Schrödinger. Minimized 3D structures of ligands were generated using Epik2.2 and within pH 7.0 +/- 2.0 in the suite. Minimization was again carried out using OPLS_2005 force field which generated 32 possible stereoisomers for each of the compounds.
Receptor Grid Generation
Grid usually restricts the active site to specific area of the receptor protein for the ligand to dock specifically within that area. In Glide, a grid was generated using default Van der Waals radius scaling factor 1.0 and charge cutoff 0.25 which was then subjected to OPLS_2005 force field for the minimized structure. A cubic box was generated around the active site (reference ligand active site) of GSK3B. Then the grid box volume was adjusted to 16×16×16 for docking to be carried out.
Glide Standard Precision (SP) Ligand Docking
SP adaptable glide docking was carried out using Glide in Maestro Schrödinger. The Van der Waals radius scaling factor and charge cutoff were set to 0.80 and 0.15 respectively for all the ligand molecules under study. Final score was assigned according to the pose of docked ligand within the active site of the receptor molecule (GSK3B). The docking result is summarized in Table 1.
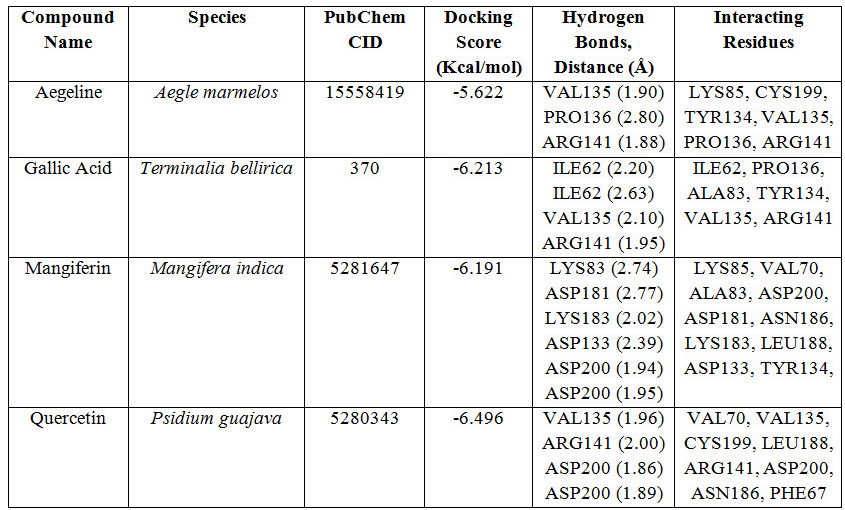
Table 1: Docking results of ligand-receptor interaction.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Ligand Based Drug Likeness Property and ADME/Toxicity Prediction
The molecular structures of every ligands were analyzed using SWISSADME server (http://www.swissadme.ch/) in order to confirm whether the ligands follow Lipinski’s rule of five or not. Physicochemical properties of ligand molecules were calculated using ORISIS property explorer (organic-chemistry.org/prog/peo/). The result of drug likeness property analysis is summarized in Table 2.
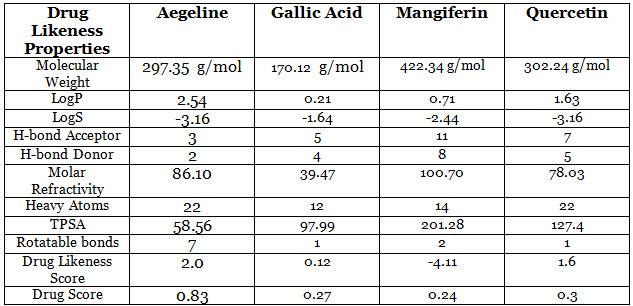
Table 2: Drug-likeness properties of selected ligand molecules.
The ADME/T for each of the ligand molecules was carried out using an online based server ADMET-SAR (http://lmmd.ecust.edu.cn/admetsar1/predict/) to predict their various pharmacokinetic and pharmacodynamic properties including blood brain barrier permeability, human abdominal adsorption, AMES toxicity, Cytochrome P (CYP) inhibitory capability, carcinogenicity, mutagenicity, Caco-2 permeability etc. The result of ADME/T for all the ligand molecules is represented in Table 3.

Table 3: ADME/T properties of selected ligand molecules.
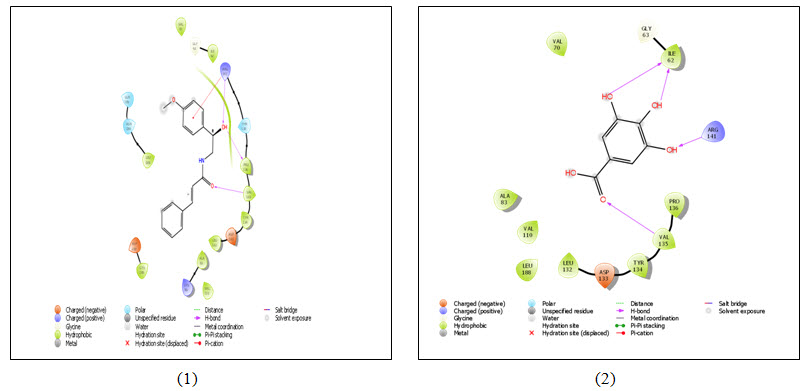
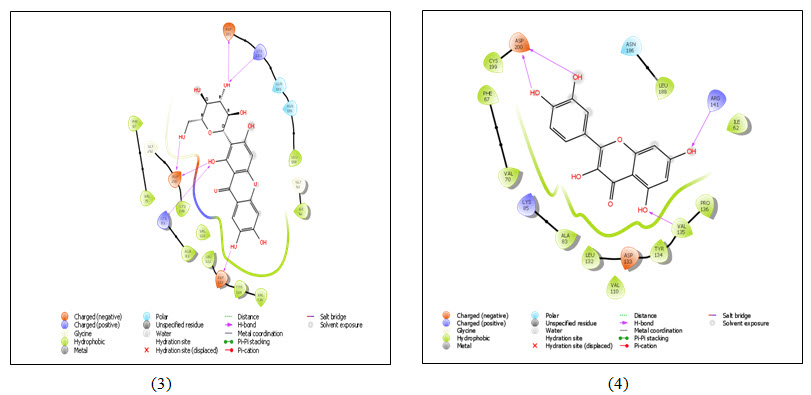
Figure 4: 2D representation of ligand-receptor interaction- (1) Aegeline; (2) Gallic acid; (3) Mangiferin and (4) Quercetin with GSK3 (PDB ID 1O9U). Colored spheres indicate amino acids and straight line between ligand and receptor represents interactions (Pink- H-bonds, Red- Pi-cation interaction). The protein pocket for the ligand is marked with the color line according to nearest atom. Interruption of line indicates opening of the pocket.
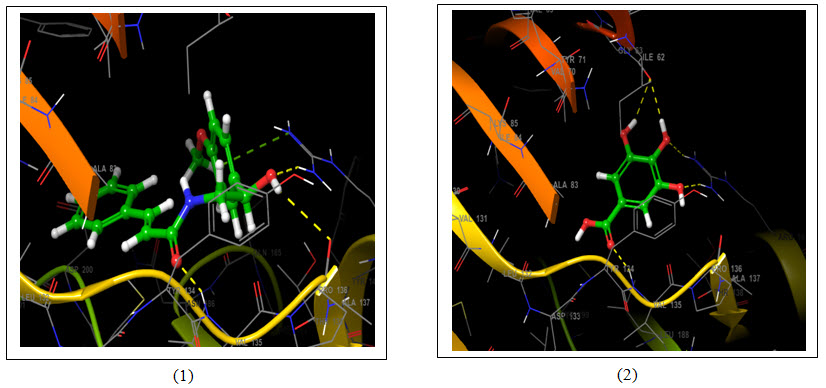
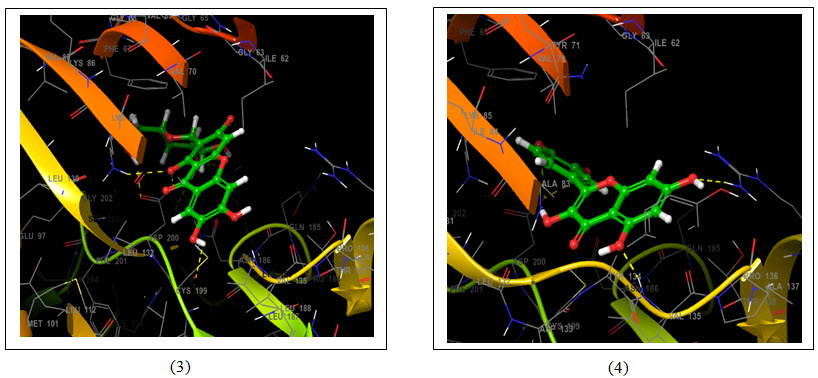
Figure 5: 3D representation of best possible pose(s) of ligands- (1) Aegeline; (2) Gallic acid; (3) Mangiferin and (4) Quercetin within the active site of the target molecule (GSK3B). Ligands are represented in green stick style and receptor is represented in ribbon style. Amino acids of target molecules are marked with three letter code. Dotted line represents interaction- Yellow- H-bonds and Green- Pi-cation interaction.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESULT
Binding Energy
Molecular docking uses specific algorithm to find the best ligand molecule that fits within the active site of a target molecule possessing best possible pose. This search algorithm utilizes an energy scoring function suggesting the ligand molecule with lowest binding energy has the highest affinity for receptor binding (Guedes et al., 2014). In this experiment all the selected ligand molecules- Aegeline, Gallic acid, Mangiferin and Quercetin interacted with the target molecule with -5.622 Kcal/mol, -6.213 Kcal/mol, -6.191 Kcal/mol and -6.496 Kcal/mol binding energy respectively (Table 1). Quercetin with the lowest binding energy has the highest affinity for GSK3B binding and the highest binding energy of Aegeline suggests lowest affinity of interaction with GSK3B. Aegeline formed hydrogen bonds with 3 amino acid residues (VAL135, PRO136 and ARG141) of the target molecule, Gallic acid formed 4 hydrogen bonds (2×ILE62, VAL135, ARG141), Mangiferin formed 6 hydrogen bonds (2×ASP200, ASP133, ASP181, LYS83, LYS183) and Quercetin formed again 4 hydrogen bonds (2×ASP200, ARG141, VAL135) as Gallic acid within the active site of GSK3B. Aegeline and Gallic acid each interacted with 6 residues within the binding pocket of the target molecules whereas Mangiferin and Quercetin interacted with 10 and 8 residues respectively.
Drug-likeness Property
Lipinski’s rule of five provides indication about the solubility, bioavailability and permeability of the drug molecule of interest. This rule assists in the shift of phase I trial to phase II trial of an investigational new drug with the help of computational approach that reduces both time and cost of the experiment (Pollastri, 2010). Aegeline, Gallic acid and Quercetin follow Lipinski’s rule of five without any violation with respect to molecular weight ( acceptable range: ≤500), number of hydrogen bond donors (acceptable range: ≤5), number of hydrogen bond acceptors (acceptable range: ≤10), lipophilicity (expressed as LogP, acceptable range: ˂5) and molar refractivity (40-130) (Table 2) (Lipinski et al., 1997). Mangiferin violates the rule of maximum H-bond donors (8) and H-bond acceptors (11). Log P value is the logarithm of partition coefficient of candidate molecule between n-octanol and water which indicates its hydrophilicity. Lower log P indicates better absorption of the drug molecule inside the cell. Log S value represents the solubility of the drug molecule and a lower value again reflects the better solubility of the candidate molecule. TPSA or topological polar surface area is again associated with absorption and permeability of the drug molecule. Any candidate with higher TSPA value often results in poor permeability in biological system. Furthermore, higher drug score and drug likeness score indicate the best possibility of a candidate molecule to have the all the physicochemical parameters within range (https://www.organic-chemistry.org/prog/peo/). Mangiferin showed the highest TSPA value (201.28) and Aegeline showed the lowest TSPA value (58.56). Gallic acid and Quercetin showed moderate level of TSPA value- 201.28 and 127.4 respectively. Quercetin exhibited better drug likeness score (1.6) and drug score (0.3) after Aegeline which exhibited likeness score of 2.0 and drug score of 0.83.
ADME/T Test
All the molecules are impermeable to blood brain barrier offering no chance to cause damage to brain (Table 3). Every molecule showed high absorption ability in intestine and didn’t show no indication of carcinogenicity. Only Gallic acid showed colorectal tissue permeability. Cytochrome P450 (CYP) family of enzymes play major roles in drug metabolism, excretion and drug-drug interaction. Inhibition of any of these enzymes may cause unusual effects like- slow degradation rate of the drug molecule, accumulation in the body and slow excretion (Newbert and Russel, 2002). Aegeline and quercetin are potent inhibitors of CYP450 3A4 and again quercetin is another inhibitor of CYP450 1A2. Gallic acid and mangiferin are not any inhibitors of any type of cytochrome P450 enzyme. Only gallic acid showed high degree of biodegradability among all the ligand molecules. Mangiferin showed the possibility to induce mutation as it positively indicated AMES toxicity.
DISCUSSION
Thousands of plants in nature have been reported to have antidiabetic activity. They have different mode of actions and deploy their effects by variety of mechanisms (Marles and Farnsworth, 1995). All the selected ligand molecules in this experiment might have antidiabetic property since all of them interacted similarly with GSK3B. However, quercetin performed the best in both docking and drug likeness property analysis and moderately well in ADME/T analysis. Mangiferin performed well than any other ligand molecules in ADME/T analysis but its violation of Lipinski’s rule may eradicate its choice as natural drug. So, quercetin could be a potent natural inhibitor of GSK3B. However, further in vivo and in vitro experiment may be required to strengthen the finding of this experiment.
CONCLUSION
Quercetin could be a greatest source of antidiabetic agent against type II diabetes since commercially available treatments are not cost effective and employs some other complications. Moreover, other three molecules also performed well indicating their potentiality to be used as antidiabetic agent. A well directed research can help us finding a sound and natural source of antidiabetic agent. Hopefully, this experiment will raise research interest among researchers about natural antidiabetic agent from medicinal plants.
REFERENCES
1. ADA (2014); Diagnosis and classification of diabetes mellitus; American Diabetes Association; Diabetes Care; 37; S81-S90.
2. Apontes P, Liu Z, Su K, Benard O, Youn DY, Li X, Li W, Mirza RH, Bastie CC, Jelicks LA, Pessin JE 2014: Mangiferin stimulates carbohydrate oxidation and protects against metabolic disorders induced by high-fat diets; Diabetes; 63(11); 3626-36.
3. Cheng FC, Shen SC, Wu JS (2009); Effect of guava (Psidium guajava L.) leaf extract on glucose uptake in rat hepatocytes; Journal of food science; 74(5); H132-8
4. Clark Jr, C.M. and Lee, D.A., (1995); Prevention and treatment of the complications of diabetes mellitus;New England journal of medicine; 332(18); 1210-1217.
5. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. (1995); Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B; Nature; 378(6559); 785.
6. Dearden, J. C. (2007); In silicoprediction of ADMET properties: how far have we come? ; Expert Opinion on Drug Metabolism & Toxicology; 3(5); 635–639.
7. Ferreira, L., dos Santos, R., Oliva, G. and Andricopulo, A., (2015); Molecular docking and structure-based drug design strategies; Molecules; 20(7); 13384-13421.
8. Grimes, C.A. and Jope, R.S., (2001); The multifaceted roles of glycogen synthase kinase 3β in cellular signaling; Progress in neurobiology; 65(4); pp.391-426.
9. Guedes IA, de Magalhães CS, Dardenne LE. (2014); Receptor–ligand molecular docking; Biophysical reviews; 6(1); 75-87.
10. Hasan T, Sultana M. (2018); Antidiabetic Potency of Bangladeshi Medicinal Plants; J Ayurvedic Herb Med; 4(1); 35-42.
11. Lipinski, C.A., Lombardo, F., Dominy, B.W. and Feeney, P.J., (1997); Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings; Advanced drug delivery reviews; 23(1-3); 3-25.
12. Marles, R.J. and Farnsworth, N.R., (1995); Antidiabetic plants and their active constituents; Phytomedicine; 2(2); 137-189.
13. Martinez, A., Castro, A., Dorronsoro, I., & Alonso, M. (2002); Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation; Medicinal Research Reviews; 22(4); 373–384
14. Morris, G.M. and Lim-Wilby, M., (2008); Molecular docking. In Molecular modeling of proteins (pp. 365-382). Humana Press.
15. Narender T, Shweta S, Tiwari P, Reddy KP, Khaliq T, Prathipati P et al. (2007); Antihyperglycemic and antidyslipidemic agent from Aegle marmelos; Bioorganic & medicinal chemistry letter 17(6); 1808-11.
16. Nebert, D.W. and Russell, D.W., (2002); Clinical importance of the cytochromes P450. ; The Lancet; 360(9340); 1155-1162.
17. Patel, S., Doble, B.W., MacAulay, K., Sinclair, E.M., Drucker, D.J. and Woodgett, J.R., (2008); Tissue-specific role of glycogen synthase kinase 3β in glucose homeostasis and insulin action; Molecular and cellular biology; 28(20); 6314-6328.
18. Pollastri, M.P., 2010. Overview on the Rule of Five. Current protocols in pharmacology, 49(1), pp.9-12.
19. Roglic G (2016); WHO Global report on diabetes: A summary; Int J Non-Commun Dis;1; 3-8
20. Sabu MC, Kuttan R (2009); Antidiabetic and antioxidant activity of Terminalia belerica. Roxb; Indian journal of experimental biology; 47(4); 270-5.
21. Shukia, R., Sharma, S.B., Puri, D., Prabhu, K.M. and Murthy, P.S., (2000); Medicinal plants for treatment of diabetes mellitus. Indian Journal of Clinical Biochemistry; 15(1);169-177.
22. Sutherland, C., Leighton, I.A. and Cohen, P., (1993); Inactivation of glycogen synthase kinase-3β by phosphorylation: new kinase connections in insulin and growth-factor signalling; Biochemical Journal; 296(1); 15-19.
23. Wang, J. and Skolnik, S., (2009); Recent advances in physicochemical and ADMET profiling in drug discovery; Chemistry & biodiversity; 6(11); 1887-1899.
24. Tapsell LC, Hemphill I, Cobiac L, Sullivan DR, Fenech M, Patch CS, Roodenrys S, Keogh JB, Clifton PM, Williams PG, Fazio VA (2006); Health benefits of herbs and spices: the past, the present, the future; The Medical Journal of Australia; 185(4); S1-S24.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








