
 About Authors:
About Authors:
Anuradha Shinde
S.R.T.M. University Nanded,
Maharashtra India
anushindebiotech@yahoo.com
ABSTRACT
The present invention relates to a filamentous fungus useful for the production of heterologous polypeptides, having been modified by recombinant DNA technology in a manner by which the expression of alkaline proteases have been completely or partially inactivated. The invention also encompasses processes for the production of proteins of interest in high yields by using the fungi of the invention. The invention furthermore relates to methods for producing such fungi and DNA constructs to be used in these methods. Extracellular alkaline proteases from Neurospora crassa is produced for the commercial requirement of proteases. The purification procedure consisted of an ammonium sulfate precipitation, dialysis, and anion-exchange chromatography, and gel filtration. We found only one enzyme identical by polyacrylamide gel electrophoresis. Optimization was done with characterization of enzyme. With a pH value of 10.0 and temperature 60 c the enzyme production was increased.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1575
INTRODUCTION TO PROTEASES
Proteases are the single class of enzymes, which occupy a pivotal position with respect to their applications in both physiological and commercial fields. They perform both degradative and synthetic functions. Proteolytic enzymes catalyze the cleavage of peptide bonds in other proteins. Advances in analytical technique have demonstrated that proteases conduct highly specific and selective modifications of proteins.
Proteases are classified on the basis of three major criteria: (1) Type of reaction catalyzed (2) Chemical nature of the catalytic site and (3) Evolutionary relationship with reference to structure. Proteases are grossly subdivided into two major groups’ i.e., exopeptides and end peptides, depending on their site of action. Based on the functional group present at the site active site, proteases are further classified into 4 prominent groups i.e. serine proteases, cysteine proteases, aspartic proteases and metallo proteases. Proteases are also divided into acid, neutral and alkaline proteases on the basis of pH range in which their activity is optimum.
Proteases occur ubiquitously in a wide diversity of sources such as plants, animals and microorganisms. Microbes are the attractive sources of proteases and have gained much popularity than any other sources because of their broad biochemical diversity. The inability of the plant and animal proteases to meet current world demands has lead to an increased interest in microbial proteases. Proteases execute a large variety of functions, extending from the cellular level to the organ and organism level. Their involvement in the life cycle of disease organisms has lead them to become a potential target for developing therapeutic agents against fatal disease such as cancer and AIDS.
Proteases have a long history of applications in different industries viz, detergents, food-brewing, meat tenderization, baking, manufacture of Soya products, debittering of protein hydrolysis’s, synthesis of aspartame, dairy, leather, silk and for recovery of silver from used x-ray films. Besides their industrial and medicinal applications, proteases play an important role in basic research. Their selective peptide bond cleavage is used in the study of sequencing of proteins.
A wide range of microorganisms including bacteria, fungi, yeast and also mammalian tissues produces alkaline proteases. The Proteolytic enzymes from Fungi are so far the most important group of enzymes produced commercially.
Technology of immobilization of enzymes was developed to extend the use of this biocatalyst for practical applications. The use of immobilized enzymes in the food, pharmaceutical and chemical industry has increased steadily during the past decade. Their use in the synthesis of chiral drugs, complex organic compounds, fine chemicals, novel specific biosensors and in bioremediation is likely to be expanded through further research and development in enzyme technology. Proteases are a complex group of enzymes, which differ in their properties. Despite the extensive research on several gaps in our knowledge of these enzymes there is still tremendous scope for improving their properties to suit projected applications.
[adsense:468x15:2204050025]
METHOD FOR EXTRACTION, PURIFICATION AND CHARECTERIZATION OF PROTEASE.
1. Screening of Neurospora for Protease Production
2. Extracellular Enzymatic activity of Microorganisms: Preparation of Crud Extract
3. Enzyme assay
4. Purification of Protease
5. Protease Assay after Purification.
6. Estimation of Standard and Purified Protease by Lowry’s Method
7. Enzyme Kinetics: Effect of PH, Temperature and Substrate on activity of Protease.
I. SCREENING OF NEUROSPORA FOR PROTEASE PRODUCTION.
Organism’s shows difference in physiological and biochemical reactions, thus by using this principle organisms were screened by providing the selective medium.
Procedure: 1. Prepare Vogel’s azocasein medium Autoclave at 1210c, 15lbs, for 15min. 2.Transfer the broth into tubes. 3. Azocasein was added after sterilization. 4.Take a loop full of pure culture and inoculate into broth.5. Place the tubes in the orbital shaking incubator for 72 hrs at30°c
Observation: -
Culture grown was checked with utility of azocasein.
II. EXTRACELLULAR ENZYMATIC ACTIVITIES OF MICROORGANISMS:
Because of their large sizes, high-molecular weight nutrients such as polysaccharides, lipids, and proteins are not capable of permeating the cell membrane. These macromolecules must first be hydrolyzed by specific extra cellular enzymes into their respective basic building blocks. These transported into the cells and used for the synthesis of protoplasmic requirements and energy production.
Preparation of Crud Extract:
After 72hours of incubation, the crude extract of the enzyme was prepared by centrifuging the culture broth at 15000rpm for 20 minutes. The supernatant was used as source of extra cellular protease. The crude enzyme extract from Vogel’s broth’s media was prepared by crushing with distilled water using mortar and pestle.
III. ENZYME ASSAY
Quantative assay of protease: Assay of Protease (by Azo-casein digest method):
Protease activity was determined with azocasein as substrate by a modified procedure described by Kreger and Lockwood (1981). For the azocasein assay method 8001.11 of the sample and400 μl of 1.5% azocasein in 0.05 M tris HCI (pH 8.5) was taken in a screw cap tube. The mixture was then incubated in a water bath at 37°C for 30 minutes. Then 2.8 ml or 5% TCA was added to stop the reaction and put on ice for 15 minutes. The solution was centrifuged at 4000 rpm for 5 minutes. In the 2 ml of supernatant 2 ml of 0.5 N NaOH solutions was added and mixed well. Absorbance was measured at 440 nm. Appropriate blank was also included. Then blank was prepared in the same manner except that 2.8 ml of TCA was added before addition of enzyme. One unit of protease activity is defined as the amount of the enzyme that produces an increase in an absorbance of 0.816 under the above assay condition.
Result:10 units of enzyme were produced per 1 ml of crude extract.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
IV. PURIFICATION OF PROTEASE: Following are the methods of purification.
a. Ammonium Sulphate Precipitation: This method is for protein purification by altering the solubility of protein (NH4)2SO4 precipitation is a simple and effective means of fractionating protein. it is based on the fact that at high salt concentration the natural tendency of protein not to aggregate is overcome, since the surface charges are neutralized Charge neutralized means that protein will tend to find together, from large complexes and hence are easy to precipitate out by mild centrifugation Since each protein will start to aggregate at a characteristic salt concentration, than approach provides a simple way of enriching for particular protein in a mixture and is used
Salting out Increase in the salt concentration implies that there is less and less water available to solubilize protein finally, protein starts to precipitate when there are not sufficient water molecules to interacts with protein molecule, then phenomenon of protein precipitation in the presence of excess salt is known as salting out
Salting In At Low concentration, the presence of salt stabilizer the various charged groups on a protein molecule, then attracting protein into the solution and enhancing the solubility of protein, then phenomenon commonly known as salting in
Fractionation: The precipitated proteins is collected and categorized to the concentration of salt solution at which it is formed. This partial collection of the separated protein pellet called Fractionation
Procedure: Protease are elute at 66% of ammonium sulfate concentration, so that we have taken 44 grams of ammonium sulfate for 100 ml of crude enzyme and then ammonium sulfate slowly added on magnetic stirrer for uniform distribution. After the addition of ammonium sulfate the crude enzyme was centrifuged at 12000 rpm for 10 minutes to collect the pellet. The pellet is collected in to the phosphate buffer pH 8.0 buffer.b. Dialysis: dialysis is the process of separating molecules in solution by the difference in their rates of diffusion through a semi permeable membrane, such as dialysis tubing.Dialysis is a common laboratory technique, and operates on the same principle as medical dialysis. Typically a solution of several types of molecules is placed into a semi permeable dialysis bag, such as a cellulose membrane with pores, and the bag is sealed. The sealed dialysis bag is placed in a container of a different solution, or pure water. Molecules small enough to pass through the tubing (often water, salts and other small molecules) tend to move into or out of the dialysis bag, in the direction of decreasing concentration. Larger molecules (often proteins, DNA, or polysaccharides) that have dimensions significantly greater than the pore diameter are retained inside the dialysis bag. One common reason for using this technique would be to remove the salt from a protein solution. The technique will not distinguish between proteins effectively.
Preparation of dialysis tube. Boil the tubing on a stir plate (preferably in the hood) in a 4L volume of 2% (w/v) sodium bicarbonate and 1mM EDTA pH 8.0.Rise the tubing in distilled water thoroughly.Boil for 10 minutes in 1mM EDTA (pH 8.0). Allow tubing to cool then store it in freezer at 4oC with tubing submerged.Before use wash out tubing with distilled water
Dialysis of Proteins After a protein has been ammonium sulfate precipitate and taken back up in buffer at a much greater protein concentration than before precipitation, the solution will contain a lot of residual ammonium sulfate which was bound to the protein. One way to remove this excess salt is to dialyze the protein against a buffer low in salt concentration.
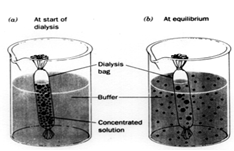
This graphic illustrates the dialysis process. First, the concentrated protein solution is placed in dialysis bag with small holes which allow water and salt to pass out of the bag while protein is retained. Next the dialysis bag is placed in a large volume of buffer and stirred for many hours (16 to 24 hours), which allow the solution inside the bag to equilibrate with the solution outside the bag with respect to salt concentration. When this process of equilibration is repeated several times (replacing the external solution with low salt solution each time), the protein solution in the bag will reach a low salt concentration:
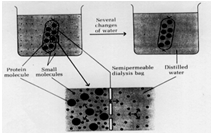
The graphic illustrates the complete dialysis process, except for it suggests you do this with distilled water. Really you want to do this process with buffer to prevent the protein from denaturing due to the fact that distilled or de ionized water is too low in salt and may have an undesirable pH for your protein, which may cause it to denature.
In fact, dialysis is a good way to exchange the buffer the protein is in at the same time you get rid of excess salt. After ammonium sulfate precipitation contains a mixture of buffers as well as excess salt. So we use the buffer we want for the next step in the purification, which is ion-exchange chromatography, as the external solution during dialysis. After the 3 step dialysis process where the protein solution is dialyzed against the starting buffer for the ion-exchange chromatography step, not only will the salt be removed but the protein will now be in the buffer needed for the next step and ready to go. Sometimes, proteins will precipitate during the dialysis process and you will need to centrifuge the solution after dialysis to remove any particles which would interfere with the next step - such as ion-exchange chromatography where particles would clog the column and prevent the chromatography step from working.
In addition, you may lose enzyme activity during dialysis. So it is a good idea to keep some of your protein solution as a sample before it is put in the dialysis bag so that it can be assayed for enzyme activity before and after dialysis.
Result: The dialyzed enzyme was collected in to the beaker and stored in refrigerator.
c. Ion Exchange chromatography. This form of chromatography relies on the attraction between oppositely charged particles. Many biological materials, for e.g. Amino acids and proteins have ionizable groups and the fact that they may carry a net positive or negative charge can be utilized in separating mixture of such a compounds. The net charge executed by these compounds is dependent on the pKa and on the pH of the solution in accordance with the Henderson Hassel Balch equation.
Ion exchange separations are carried out mainly in columns packed with an ion-exchanger. There are two types of ion exchanger, namely, Cation and Anion Exchangers.Cation exchangers possess negatively charged groups and these will attract positively charged cations. These exchangers are also called acidic ion exchange materials because their negative charges result from the ionization of acidic groups. Carboxy methyl cellulose is a cat ion exchanger; here CM-cellulose is most applicable for the separation of proteins which are positively charged at around pH 4-5.Anion exchangers have positively charged groups that will attract negatively charged anions. The term basic ion exchange material is also used to describe this exchanger, as positive charges generally results from the association of the protons with basic groups.
The most frequently used are Diethylaminoethyl (DEAE)-cellulose which is anion exchanger. The DEAE group, -CH2H5NH(C2H5)2,is highly positive charge at pH 6-8, so DEAE- cellulose is most useful for the chromatography of protein which are negatively charged in this range.Elution of the proteins from the columns may be brought about by changes in either salt concentration of salt (e.g. NaCl) increases; protein is displaced from DEAE by Cl- ions and from CMC-cellulose by the cat ion Na+
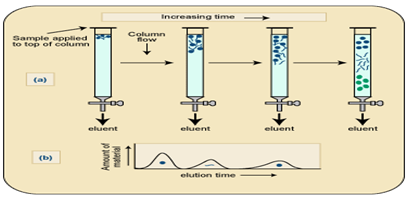
Buffer preparation: 1. Add 2.78gms in 100ml sodium phosphate solution and it was taken as X-solution. 2. Then add 0.2M dibasic sodium phosphate 5.365mg in 100ml water and it was taken as Y-solution. 3.From solution X take 94.5 ml and from solution Y we took 4.5ml in a conical flask and it was makeup to 200ml.
Preparation of chemicals: Phosphate buffer, NaCl
|
Sl.No |
TEST TUBE |
BUFFER in ml |
NACL in ml |
Distilled water in ml |
OD At 660nm |
|
1 |
25mm |
0.25ml |
0.125 |
4.625 |
0.10 |
|
2 |
50mm |
0.25 |
4.5 |
0.55 |
|
|
3 |
75mm |
0.375 |
4.375 |
0.05 |
|
|
4 |
100mm |
0.5 |
4.15 |
0.08 |
|
|
5 |
125mm |
0.625 |
4.125 |
0.15 |
|
|
6 |
150mm |
0.75 |
4.0 |
0.12 |
|
|
7 |
175mm |
0.875 |
3.875 |
0.10 |
|
|
8 |
200mm |
1 |
3.75 |
0.08 |
|
|
9 |
225mm |
1.125 |
3.5 |
0.00 |
|
|
10 |
250mm |
1.25 |
3.25 |
0.00 |
Procedure: First add phosphate buffer to clean the column, then add the protein sample, Then add the serial sample test tubes, Then take out the purified protein sample.
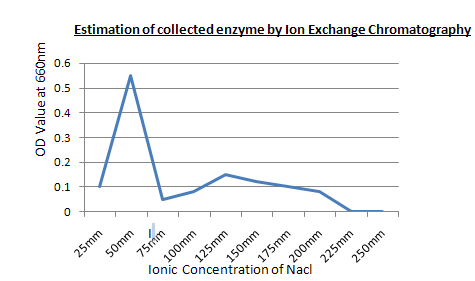
Result: The purified protein sample is collected at 50 mM concentrated NaCl.
V. PROTEASE ASSAY AFTER PURIFICATION:
Aromatic amino acids absorb (due to their benzene rings) resonance at 280nm. The absorbance is a measure of liberation of tyrosine. The activity of protease liberates tyrosine after proteolytic cleavage. In order to ensure free hydrolysis and thereby facilitate proteolytic cleavage, the substrate and the test solution are subjected to digestion by TCA.
Reagents: 0.5%BSA solution,50mM Tris HCl (pH 8.0), 10%trichloroacetic acid.
Procedure:
To 0.5 ml of 0.5% of BSA solution made up in 50mM tris HCl, adds 0.1ml of enzyme solution and 0.75ml of 10%trichloro acetic acid and incubated at 370C for 20 min. After incubation, add 0.5ml of 0.5%BSA solution to the blank and 0.75ml of 10% trichloro acetic acid to the test and left at 37oC for 20 min. Centrifuged at 10,000rpm for 20 min to separate the supernatant. The absorbance of the blank and test was read at 280nm and increase in the absorbance at this wavelength indicates the enzyme activity.
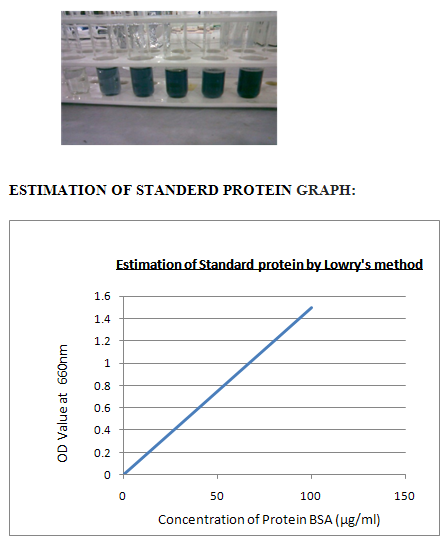
VI. ESTIMATION STANDARD PROTIEN BY LOWRY’S METHOD:
|
S.No |
Vol. of std.(BSA) |
Vol. of D/W |
Concentration of BSA in µg/ml |
Vol. of Alkaline copper sulphate reagent |
Incubation at room temperature |
Vol of F-c Reagent |
Boiling at 100 c in water bath |
O.D values at 660nm |
|
Blank |
0.0ml |
1ml |
00 |
5ml |
10min |
0.5 |
10min |
0.00 |
|
1. |
0.2ml |
0.8ml |
20 |
5ml |
10min |
0.5 |
10min |
0.30 |
|
2. |
0.4ml |
0.6ml |
40 |
5ml |
10min |
0.5 |
10min |
0.60 |
|
3. |
0.6ml |
0.4ml |
60 |
5ml |
10min |
0.5 |
10min |
0.90 |
|
4. |
0.8ml |
0.2ml |
80 |
5ml |
10min |
0.5 |
10min |
1.20 |
|
5. |
1.0ml |
0.0ml |
100 |
5ml |
10min |
0.5 |
10min |
1.50 |
Procedure:1. Take a series of test tubes and label them as 1 to 5. 2.Then add different concentrations of standard 0.2 to 1.00 to the test tubes. 3.Then add 5 ml of alkaline copper sulphate. 4.Incubate the test tubes for 10 minutes at room temperature. 5.Then add 0.5ml of FC reagent to all the test tubes.6. Then keep the test tubes in hot water bath at 100 c for 10 minutes.
7.Maintain the tube, blank as same as, instead of standard add distilled water to adjust the calorimeter to zero. 8. Then cool the test tubes and add 1 ml ethanol all the test tubes. 9. Then take O.D values by using calorimeter at 660nms 10. Then plot the graph between concentration Vs O.D values.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
OBSERVATION:
Estimation of purified enzyme:
|
S.No |
Vol. of std.(BSA) |
Vol. of D/W |
Concentration of Purifier Enzyme in µg/ml |
Vol. of Alkaline copper sulphate reagent |
Incubation at room temperature |
Vol of Fc Reagent |
Boiling at 1000C in water bath |
O.D values at 660nm |
|
Blank |
0.0ml |
1.0ml |
00 µg |
5ml |
10min |
0.5ml |
10min |
0.00 |
|
1. |
0.2ml |
0.8ml |
20 µg |
5ml |
10min |
0.5ml |
10min |
0.20 |
|
2. |
0.4ml |
0.6ml |
40 µg |
5ml |
10min |
0.5ml |
10min |
0.40 |
|
3. |
0.6ml |
0.4ml |
60 µg |
5ml |
10min |
0.5ml |
10min |
0.60 |
|
4. |
0.8ml |
0.2ml |
80 µg |
5ml |
10min |
0.5ml |
10min |
0.80 |
|
5. |
1.0ml |
0.0ml |
100 µg |
5ml |
10min |
0.5ml |
10min |
1.00 |
|
6. |
Unknown 0.5 ml |
0.5ml |
60 µg |
5ml |
10min |
0.5ml |
10min |
0.60 |
Observation:

ESTIMATION OF PURIFIED ENZYME
Graph:
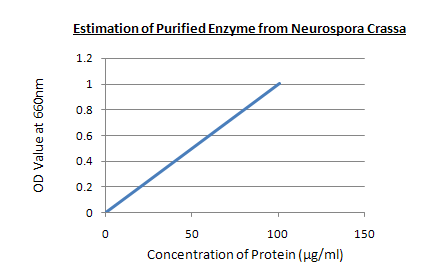
RESULT: In 0.5 ml of purified enzyme solution the concentration of enzyme is 60µg which is taken as unknown sample.
VII. OPTIMIZATION:
The activity of enzyme is influenced by various factors includes pH, temperature and Substrate.
EFFECT OF pH: The rate of enzymatic reaction depends on pH of medium. The enzymatic activity is maximum at a particular pH called optimum pH.
Reagents: enzyme, Phosphate Buffer, 0.5% BSA, TCA, FCR, 5% sodium hydroxide
Procedure: 1.0 ml of phosphate buffer of different pH 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0 and 12.0 was taken in nine test tubes. 1ml of 0.5%BSA was added to all the test tubes followed by the addition of 1ml of enzyme.All the test tubes were incubated at 370C for 10min. After incubation, add 2.0 ml of 10% TCA was added and then the contents of the tubes were centrifuged at 10,000rpm for 10min. To the 0.5ml of the supernatant, 2ml of 5% sodium hydroxide and 0.5 ml of FCR were added and incubated at 370 C for 10 min. After incubation, the enzyme activity was measured at 660nm.
|
Sr. No |
Buffer of Various PH |
Vol of BSA 0.5% |
Vol of Enzyme |
Incubation at 370C |
Vol of TCA |
Vol of NaoH |
Incubation For 10min at 370C |
Centrifuge for 10min at 10000 |
OD At 660nm |
|
1 |
4ml |
1ml |
1ml |
10min |
2ml |
2ml |
10min |
0.5 |
0.04 |
|
2 |
5ml |
1ml |
1ml |
10min |
2ml |
2ml |
10min |
0.5 |
0.09 |
|
3 |
6ml |
1ml |
1ml |
10min |
2ml |
2ml |
10min |
0.5 |








