
ABOUT AUTHORS:
Sweety Sharma*, Amardeep Ankalgi, Chandra Shekhar Sharma, Hemendra Pratap Singh, Priyadarshani Kamble, Mahendra Singh Ranawat.
Deptt. Of Pharmaceutical Chemistry, B.N.College of Pharmacy,
Udaipur-313002, Rajasthan, India.
*sweet2pharma@gmail.com
ABSTRCT
Impurity is defined as any substance coexisting with the original drug, such as starting material or intermediates or that is formed, due to any side reactions. According to the International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) guideline on impurities in new drug substances, an impurity is defined as 'any component of the new drug substance that is not the chemical entity defined as the new drug substance’. Identification of impurities is done by variety of Chromatographic and Spectroscopic techniques, either alone or in combination with other techniques. There are different methods for detecting and characterizing impurities with TLC, HPLC, and HPTLC etc. The most exploited techniques, for impurity profiling of drugs are LC-MS-MS, LC-NMR, LCNMR- MS, GC-MS, and LC-MS.
REFERENCE ID: PHARMATUTOR-ART-1786
1. INTRODUCTION
Group of analytical activity, the aim of which is the detection, identification, or structure elucidation & quantitative determination of organic & inorganic impurities, as well as residual solvents in bulk drug & formulation [1]. A description of the identified and unidentified impurities present in a new drug substance[2]. Drug impurity profiling, i.e. identification, structure elucidation and quantitative determination of impurities and degradation products in bulk drug materials and pharmaceutical formulations[3]. Pharmaceuticals impurities are the unwanted chemicals that remain with the active pharmaceutical ingredients (APIs) or are developed during formulation or upon aging of both API and formulated APIs to medicines [6]. According to the International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) guideline on impurities in new drug substances,[1] an impurity is defined as 'any component of the new drug substance that is not the chemical entity defined as the new drug substance'.[4]
2. OBJECTIVES [7]
It provides useful information for drug low enforcement authorities. The practical value of this studies for routine analysis is low enforcement investigate work in four different areas as following-
1) Establishing specific links between two or more samples.
2) Establishing drug distribution patterns.
3) Identifying the source of drug sample.
4) Monitoring methods used for drug manufacturing.
Depending on the nature of the drug sample investigated, the information generated by this method may be used to identify from where, how & to what extent the drug has been distributed. Enables measurement of the relative concentration of major, minor & trace elements. By such an approach, a characteristic chemical signature can be assigned to every drug sample.
3. ACCEPTANCE CRITERIA [8,9, 10, 11]
The new drug substance specification should include, where applicable, the following list of impurities:
? Each specified identified impurity;
? each specified unidentified impurity;
? Any unspecified impurity with an acceptance criterion of not more than (≤) the identification threshold;
? total impurities.
|
Maximum Daily Dose1 |
Reporting Threshold2,3 |
Identification Threshold3 |
Qualification Threshold3 |
|
≤ 2g/day |
0.05% |
0.10% or 1.0 mg per day intake (whichever is lower) |
0.15% or 1.0 mg per day intake (whichever is lower) |
|
> 2g/day |
0.03% |
0.05% |
0.05% |
4. TYPES OF IMPURITY
4.1 ACCORDING TO USP [8, 14]
The United States Pharmacopoeia (USP) classifies impurities in various sections (A) Impurities in Official Articles (B) Ordinary Impurities. This found in bulk pharmaceutical chemicals that are innocuous by virtue of having no significance on biological activity of the drug substance. These impurities may arise out of the synthesis, preparation or degradation of chemical.And (C) Organic Volatile Impurities-Organic volatile chemicals are produced in the Manufacture of drug substances or excipients or the preparation of drug products; they are volatile in nature and by themselves get removed out at time of storage or processing.
4.2 ACCORDING TO ICH GUIDELINES [9, 10, 11]
The new drug substance specifications should include, limits for-
i) Organic Impurities
- Each specific unidentified impurity at or above 0.1%
- Any unspecific impurity, with limit of not more than 0.1%
- Total impurities
ii) Residual solvents
iii) Inorganic impurities
4.3 ACCORDING TO LITERATURE [12, 13]
4.3.1 Organic impurities
They are the most common impurities found in every API unless proper care is taken throughout the multistep synthesis. Although the end products are always washed with solvents, there is always a chance that the residual unreacted starting material remain, unless the manufactures are very careful about the impurities.
It can be any of following.
a. Starting Material-Example: In PCM Bulk, there is a limit test for p-aminophenol, which could be starting material or intermediate for synthesis.
b. By product-Example: In the case of paracetamol bulk, diacetylated+ paracetamol may be formed as a by product.
c. Intermediates
d. reagents
The spectroscopic studies (NMR, IR, MS etc. ) conducted to characterize the structure of actual impurities present in the drug substance above an apparent level of 0.1% (e.g., calculated using the response factor of the drug substance) should be described. All recurring impurities above an apparent level of 0.1% in batches manufactured by the proposed commercial process should be identified of these studies.
4.3.2 Inorganic impurities
They may also derive from the manufacturing processes used for bulk drugs. They are normally known & identified & include the Reagents, Ligands, Catalysts, Heavy Metals, Filter aids, Charcoals etc. Inorganic impurities are normally detected and quantified using Pharmacopeial or other appropriate standards. Carryover of catalysts to the drug substance should be evaluated during development.
4.3.3 Residual solvents
Residual solvents are organic volatile chemicals used during the manufacturing processes or generated during the production. Some solvents that are known to cause toxicity should be avoided in the production of the drugs.
Depending on the possible risk to humans, residual solvents are divided into 3 classes,
Class 1: Human carcinogens.
Class 2: Non genotoxic.
Class 3: Lower risk to human health.
4.3.4 Genotoxic impurities
These are the impurities that damage DNA by mutation of genatic code. Example: Alkylation
5. FACTORS AFFECTING IMPURITY [3, 16, 17]
5.1 Crystallization
Based on the realization that the nature of structure adopted by a given compound upon crystallization could exert a profound effect on the solid-state properties of that system, the pharmaceutical industry is required to take a strong interest in polymorphism and solvatomorphism as per the regulations laid down by the regulatory authorities. Polymorphism is the term used to indicate crystal system where substances can exist in different crystal packing arrangements, all of which have the same elemental composition. Whereas, when the substance exists in different crystal packing arrangements, with a different elemental composition; the phenomenon is known as Solvatomorphism.
5.2 Stereochemistry
Stereochemistry related compounds; that is, those compounds that have similar chemical structure but different spatial orientation, these compounds can be considered as impurities in the API’s. Chiral molecules are frequently called enantiomers. The single enantiomeric form of chiral drug is now considered as an improved chemical entity that may offer a better pharmacological profile and an increased therapeutic index with a more favorable adverse reaction profile.
5.3 Synthetic Intermediates & By Product
Impurities in pharmaceutical compounds or a new chemical entity (NCE) can originate during the synthetic process from raw materials, intermediates and/or by-products. For example, impurity profiling of ecstasy tablets by GC-MS, and MDMA samples, produced impurities in intermediates via reductive amination route.
5.4 Residual Solvents
Residual solvents are organic volatile chemicals used during the manufacturing process or generated during the production as vehicle, dissolution media or for granulation. Some solvents that are known to cause toxicity should be avoided in the production of bulk drugs. A selective gas chromatography (GC) method has been developed to determine the purity of acetone, dichloromethane, methanol and toluene. Using this method, the main contaminants of each organic solvent can be quantified.
5.5 Formulation Related Impurities
Many impurities in a drug product can originate from excipients used to formulate a drug substance. In addition, a drug substance is subjected to a variety of conditions in the process of formulation that can cause its degradation or have other undesirable reactions.Solutions and suspensions are inherently prone to degradation due to hydrolysis or solvolysis. Fluocinonide Topical Solution USP, 0.05%, in 60-mL bottles, was recalled in the United States because of degradation/impurities leading to sub- potency. In general, liquid dosage forms are susceptible to both degradation and microbiological contamination. Microbiological growth resulting from the growth of bacteria, fungi, and yeast in a humid and warm environment may results in unsuitability of an oral liquid product for safe human consumption.
5.6 Impurities During Storage
A number of impurities can originate during storage or shipment of drug products. It is essential to carry out stability studies to predict, evaluate, and ensure drug product safety.
5.7 Method Related Impurities
Due to deviation in pH and column temperature. Example:The intramolecular cyclic reaction of diclofenac sodium forming an indolinone derivative and sodium hydroxide. The formation of this impurity has been found to depend on initial pH of the formulation.
5.8 Environmental
Effect of humidity, temperature, light.
5.9 Mutual Interaction
Most vitamins are very labile and on aging they create a problem of instability in different dosage forms, especially in liquid dosage forms. Degradation of vitamins does not give toxic impurities; however, potency of active ingredients drops below Pharmacopoeial specifications.
5.10 Functional group
Ester hydrolysis can be explained with a few drugs viz aspirin, benzocaine, cefotaxime, ethyl paraben, and cefpodoxime proxetil.
Hydrolysis is the common phenomenon for ester type of drugs, especially in liquid dosage forms viz benzylpenicillin, oxazepam and lincomycin.
Oxidative degradation of drugs like hydrocortisone, methotrexate, hydroxyl group directly bonded to an aromatic ring (viz phenol derivatives such as catecholamines and morphine), conjugated dienes (viz vitamin A and unsaturated free fatty acids), heterocyclic aromatic rings, nitroso and nitrite derivatives, and aldehydes (especially flavorings) are all susceptible to oxidative degradation.
6. GENERAL SCHEME FOR DRUG IMPURITY PROFILING [1]
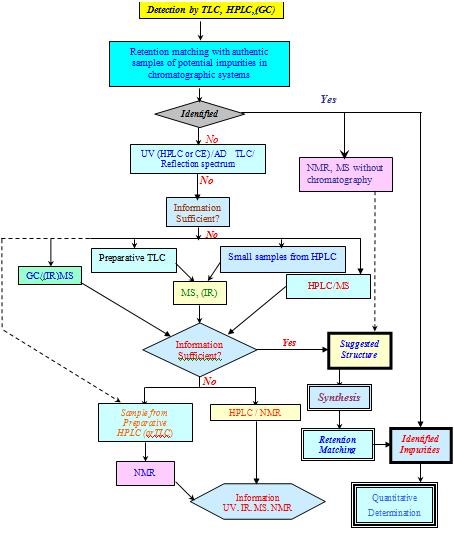
7. ISOLATION TECHNIQUES [18-22]
Isolation Methods It is often necessary to isolate impurities. But if the instrumental methods are used, isolation of impurities is avoided as it directly characterizes the impurities.
Generally, chromatographic and non-chromatographic techniques are used for isolation of impurities prior its characterization. The term ‘chromatographic reactor’ refers to the use of an analytical-scale column as both a flow-through reactor, and simultaneously, as separation medium for the reactant(s) and product(s). By using an HPLC, chromatographic reactor approach, the solution-phase hydrolysis kinetics of the Aprepitant (EmendTM) prodrug, fosaprepitant dimeglumine, were investigated. In loratidine, impurity found was ofloratidine; other examples include celecoxib, and amikacin. A list of methods that can be used for isolation of impurities is given below.
7.1 Solid-Phase Extraction Methods
Solid phase extraction (SPE) is an increasingly useful sample preparation technique. With SPE, many of the problems associated with liquid – liquid extraction can be prevented, such as incomplete phase separation, less-than-quantitative recoveries, use of expensive, breakable specialty glassware, and disposal of large quantities of organic solvents. SPE is more efficient than liquid – liquid extraction, yields quantitative extractions that are easy to perform, is rapid, and can be automated. Solvent use and laboratory time are reduced. SPE is used very often to prepare liquid samples and extract semi-volatile or nonvolatile analytes, and can also be used with solids that are pre-extracted into solvents. SPE products are excellent for sample extraction, concentration, and cleanup. They are available in a wide variety of chemistries, adsorbents, and sizes. Selecting the most suitable product for each application and sample is important.
7.2 Liquid – Liquid Extraction Methods
Liquid – liquid extraction, also known as solvent extraction and partitioning, is a method to separate compounds based on their relative solubilities in two different immiscible liquids, usually water and an organic solvent. It is an extraction of a substance from one liquid phase into another liquid phase. Liquid – liquid extraction is a basic technique in chemical laboratories, where it is performed using a separating funnel. This type of process is commonly performed after a chemical reaction as part of the workup.
7.3 Accelerated Solvent Extraction Methods
Accelerated Solvent Extraction (ASE) is a better technique for the extraction of solid and semisolid sample matrices, using common solvents, at elevated temperatures and pressures. ASE systems are available in the entry level ASE 150 system and the fully automated ASE 350. Extractions that normally take hours can be done in minutes using ASE with pH hardened pathways, using Dionium™ components. Compared to techniques such as Soxhlet and sonication, ASE generates results in a fraction of the time. The many steps involved in sample preparation can now be automated with the ASE flow-through technology. Filtration and clean up of solid samples can be achieved as part of the solvent extraction process in a single step. ASE offers a lower cost per sample than other techniques, reducing solvent consumption by up to 90%.
7.4 Supercritical Fluid Extraction
Supercritical Fluid Extraction (SFE) is the process of separating one component (the extractant) from another (the matrix), using supercritical fluids as the extracting solvent. Extraction is usually from a solid matrix, but can also be from liquids. SFE can be used as a sample preparation step for analytical purposes, or on a larger scale to either strip unwanted material from a product (e.g., decaffeination) or collect a desired product (e.g., essential oils). Carbon dioxide (CO2) is the most used supercritical fluid, sometimes modified by co-solvents such as ethanol or methanol. Extraction conditions for supercritical CO2 are above the critical temperature of 31°C and critical pressure of 72 bars. Addition of modifiers may slightly alter this.
7.5 Column Chromatography
Column chromatography in chemistry is a method used to purify individual chemical compounds from mixtures of compounds. It is often used for preparative applications on scales from micrograms to kilograms. The classical preparative chromatography column is a glass tube with a diameter of 50 mm and a height of 50 cm to 1 m with a tap at the bottom. Two methods are generally used to prepare a column; the dry method and the wet method. The individual components are retained by the stationary phase differently and separate from each other while they are running at different speeds through the column with the eluent. At the end of the column they elute one at a time. During the entire chromatography process the eluent is collected in a series of fractions. The composition of the eluent flow can be monitored and each fraction is analyzed for dissolved compounds, for example, by analytical chromatography, UV absorption or fluorescence. Colored compounds (or fluorescent compounds, with the aid of an UV lamp) can be seen through the glass wall as moving bands.
7.6 Flash Chromatography
Distillation, re-crystallization, and extraction are all important techniques for the purification of organic compounds. However, the technique used most commonly in modern organic research is ‘flash’ chromatography. In traditional column chromatography the sample to be purified is placed on top of a column containing some solid support, often silica gel. The rest of the column is then filled with a solvent (or a mixture of solvents), which then runs through the solid support under the force of gravity. The various components to be separated travel through the column at different rates and are then collected separately as they emerge from the bottom of the column. Unfortunately, the rate at which the solvent percolates through the column is slow. In flash chromatography, however, air pressure is used to speed up the flow of the solvent, dramatically decreasing the time needed to purify the sample.
7.7 Thin Layer Chromatography
Thin layer chromatography (TLC) is a chromatography technique used to separate mixtures. Thin layer chromatography is performed on a sheet of glass, plastic or aluminum foil, which is coated with a thin layer of adsorbent material, usually silica gel, aluminium oxide or cellulose. This layer of adsorbent is known as the stationary phase.
After the sample has been applied on the plate, a solvent or solvent mixture (known as the mobile phase) is drawn up the plate via capillary action. As different analytes ascend the TLC plate at different rates, separation is achieved.
Thin layer chromatography finds many applications to determine the components that are contained in plants. It is also used for monitoring organic reactions and analyzing ceramides and fatty acids; for the detection of pesticides or insecticides in food and water; for analyzing the dye composition of fibers in forensics and identifying compounds present in a given substance, and for assaying the radiochemical purity of radio pharmaceuticals. A number of enhancements can be made to the original method, to automate the different steps, to increase the resolution achieved with TLC, and to allow more accurate quantization. This method is referred to as HPTLC or ‘high performance TLC’.
7.8 Supercritical Fluid Chromatography
Supercritical Fluid Chromatography (SFC) is a form of normal phase chromatography that is used for the analysis and purification of low-to-moderate molecular weight, thermally labile molecules. It can also be used for the separation of chiral compounds. Its principles are similar to those of HPLC, however SFC typically utilizes carbon dioxide as the mobile phase; therefore, the entire chromatographic flow path must be pressurized.
7.9 Gas Chromatography
Gas–liquid chromatography (GLC) or simply gas chromatography (GC), is a common type of chromatography used in analytical chemistry for separating and analyzing compounds that can be vaporized without decomposition. Typical uses of GC include testing the purity of a particular substance or separating the different components of a mixture (the relative amounts of such components can also be determined). In some situations, GC may help in identifying a compound. In preparative chromatography, GC can be used to prepare pure compounds from a mixture.
7.10 High Performance Liquid Chromatography
High performance liquid chromatography (or high pressure liquid chromatography, HPLC) is a form of column chromatography used frequently in biochemistry and analytical chemistry, to separate, identify, and quantify compounds, based on their idiosyncratic polarities and interactions with the column's stationary phase. HPLC utilizes different types of stationary phases (typically, hydrophobic saturated carbon chains), a pump that moves the mobile phase(s) and analyte through the column, and a detector that provides a characteristic retention time for the analyte. The detector may also provide other characteristic information (i.e., UV / Vis spectroscopic data for the analyte if so equipped). Analyte retention time varies depending on the strength of its interactions with the stationary phase, the ratio / composition of the solvent(s) used, and the flow rate of the mobile phase.
7.11 Gas Chromatography – Mass Spectroscopy
Gas chromatography – mass spectrometry (GC – MS) is a method that combines the features of gas – liquid chromatography and mass spectrometry, to identify different substances within a test sample. Applications of GC – MS include drug detection, fire investigation, environmental analysis, explosives investigation, and identification of unknown samples. Additionally, it can identify trace elements in materials that were previously thought to have disintegrated beyond identification. The GC – MS has been widely heralded as a ‘gold standard’ for forensic substance identification because it is used to perform a specific test. A specific test positively identifies the actual presence of a particular substance in a given sample. A non-specific test merely indicates that a substance falls into a category of substances. Although a non-specific test could statistically suggest the identity of the substance, this could lead to false positive identification.
7.12 Gravimetric Analysis
Gravimetric analysis describes a set of methods in analytical chemistry for the quantitative determination of an analyte based on the mass of a solid. A simple example is the measurement of solids suspended in a water sample: A known volume of water is filtered, and the collected solids are weighed. In most cases, the analyte must first be converted to a solid by precipitation, with an appropriate reagent. The precipitate can then be collected by filtration, washed, dried to remove traces of moisture from the solution, and weighed. The amount of analyte in the original sample can then be calculated from the mass of the precipitate and its chemical composition.
7.13 UV Spectrometry
Ultraviolet (UV) spectroscopy is a physical technique of the optical spectroscopy that uses light in the visible, ultraviolet, and near infrared ranges. The Beer-Lambert law states that the absorbance of a solution is directly proportional to the concentration of the absorbing species in the solution and the path length. Thus, for a fixed path length, UV / VIS spectroscopy can be used to determine the concentration of the absorber in a solution. It is necessary to know how rapidly the absorbance changes with concentration.
7.14 Infrared Spectroscopy
Infrared spectroscopy is the subset of spectroscopy that deals with the infrared region of the electromagnetic spectrum. It covers a range of techniques, the most common being a form of absorption spectroscopy. As with all spectroscopic techniques, it can be used to identify compounds and investigate sample compositions. A common laboratory instrument that uses this technique is an infrared spectrophotometer. The infrared portion of the electromagnetic spectrum is usually divided into three regions; the near-, mid- and far-infrared, named according to their relation to the visible spectrum. The far-infrared, approximately 400 – 10 cm-1 (1000 – 30 μm), lying adjacent to the microwave region, has low energy and may be used for rotational spectroscopy. The mid-infrared, approximately 4000 – 400 cm-1 (30 – 2.5 μm), may be used to study the fundamental vibrations and associated rotational-vibrational structure.
7.15 Fluorescence Spectroscopy
Fluorescence spectroscopy is called as fluorometry or spectrofluorometry. It is a type of electromagnetic spectroscopy, which analyzes the fluorescence from a sample. It involves using a beam of light, usually ultraviolet light, which excites the electrons in the molecules of certain compounds and causes them to emit light of a lower energy, typically, but not necessarily, visible light.
7.16 Mass spectrometry
Mass spectrometers are used in the industry and academia for both routine and research purposes. The following list is just a brief summary of the major mass spectrometric applications:
Biotechnology: Is the analysis of proteins, peptides, and oligonucleotides.
Pharmaceuticals: Deal with drug discovery, combinatorial chemistry, pharmacokinetics, and drug metabolism.
Clinical: Deals with neonatal screening, hemoglobin analysis, and drug testing.
Environmental: Deals with polycyclic aromatic hydrocarbons (PAHs), Polychlorinated biphenyls (PCBs), water quality, and food contamination.
Geological: Deals with the oil composition.
The instruments include: Ionization source, for example, electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI); Analyzer mass to charge (m/z), for example, quadruple and magnet; FT-ICR detector, for example, the photomultiplier micro-channel plate electron multiplier.
Ionization methods include the following:
Atmospheric Pressure Chemical Ionization (APCI), Chemical Ionization (CI), Electron Impact (EI), Electrospray ionization (ESI), Fast atom bombardment (FAB), Field desorption / Field ionization (FD / FI), Matrix-assisted laser desorption ionization (MALDI), and Thermospray ionization (TSP).
7.17 NMR spectroscopy
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful and theoretically complex analytical tool. In NMR, the chemical environment of the specific nuclei is deduced from the information obtained about the nuclei.
Nuclear magnetic resonance (NMR) is a property that magnetic nuclei have in a magnetic field and the applied electromagnetic (EM) pulse or pulses, which cause the nuclei to absorb energy from the EM pulse and radiate this energy back out. The energy radiated back out is at a specific resonance frequency, which depends on the strength of the magnetic field and other factors. This allows for the observation of specific quantum mechanical magnetic properties of an atomic nucleus. Many scientific techniques exploit NMR phenomena, to study molecular physics, crystals, and non-crystalline materials, through NMR spectroscopy. NMR is also routinely used in advanced medical imaging techniques, such as in magnetic resonance imaging (MRI).
All stable nuclides that contain an odd number of protons and / or neutrons (see Isotope) have an intrinsic magnetic moment and angular momentum, in other words a spin > 0, while all nuclides with even numbers of both have spin 0. The most commonly studied nuclei are 1H (the most NMR-sensitive isotope after the radioactive 3H) and 13C, although nuclei from isotopes of many other elements (e.g.,2H, 10B, 11B, 14N, 15N, 17O, 19F, 23Na, 29Si, 31P, 35Cl, 113Cd, and 195Pt) are studied by high-field NMR spectroscopy as well.
A key feature of NMR is that the resonance frequency of a particular substance is directly proportional to the strength of the applied magnetic field. It is this feature that is exploited in imaging techniques. If a sample is placed in a nonuniform magnetic field, then the resonance frequencies of the sample's nuclei depend on where in the field they are located. As the resolution of the imaging techniques depends on how big the gradient of the field is, many efforts are made to develop more powerful magnets, often using superconductors. The effectiveness of NMR can also be improved using hyper polarization, and / or two-dimensional, three-dimensional, and higher dimension multi-frequency techniques.
Nuclear Magnetic Resonance phenomena are also utilized in low-field NMR, NMR spectroscopy, and MRI in the Earth's magnetic field (referred to as Earth's field NMR).
7.18Raman spectroscopy
Raman spectroscopy is a spectroscopic technique used to study vibrational, rotational, and other low-frequency modes in a system. It relies on the inelastic scattering or the Raman scattering of the monochromatic light, usually from a laser, in the visible, near infrared, or near ultraviolet range. The laser light interacts with the photons or other excitations in the system, resulting in the energy of the laser photons being shifted up or down. The shift in energy gives information about the phonon modes in the system. Infrared spectroscopy yields similar, but complementary, information. Typically, a sample is illuminated with a laser beam. Light from the illuminated spot is collected with a lens and sent through a monochromator. Wavelengths close to the laser line, due to elastic Rayleigh scattering, are filtered out, while the rest of the collected light is dispersed onto a detector.
Spontaneous Raman scattering is typically very weak, and as a result the main difficulty of Raman spectroscopy is separating the weak inelastically scattered light from the intense Rayleigh scattered laser light. Modern instrumentation almost universally employs notch or edge filters for laser rejection and spectrographs (either axial transmissive (AT) or Czerny-Turner (CT) monochromator) or FT (Fourier transform spectroscopy), and CCD detectors.
There are a number of advanced types of Raman spectroscopy, including surface-enhanced Raman, tip-enhanced Raman, polarized Raman, stimulated Raman (analogous to stimulated emission), transmission Raman, spatially-offset Raman, and hyper Raman. Heavily B-doped polycrystalline diamond films ([B] ≥1019 cm-3) are studied by Raman spectroscopy and electron spin resonance. The formation of an impurity band is accompanied by Fano-type interference for the one-photon scattering. Bands at 1200 and 500 cm-1 are observed in Raman spectroscopy for concentrations above 1020 cm-3. They are related to the maxima in the photon density of states, and are ascribed to disordered regions or crystalline regions of very small size. The concentration of defects associated with the paramagnetic signal observed around g = 2.0030 increases drastically above 1021B cm-3. The Mott insulator-metal transition is accompanied by the presence of a new paramagnetic signal (g = 2.0007 for 2 × 1020B cm-3, g = 1.9990 for 1021B cm-3) ascribed to free holes in the impurity band.
8. Applications
Numerous applications have been sought in the areas of drug designing and in monitoring quality, stability, and safety of pharmaceutical compounds, whether produced synthetically, extracted from natural products or produced by recombinant methods. The applications include alkaloids, amines, amino acids, analgesics, antibacterials, anticonvulsants, antidepressant, tranquilizers, antineoplastic agents, local anesthetics, macromolecules, steroids, miscellaneous 23
Table 1 Goals of impurity investigations
|
Process-related impurities |
Degradation–related impurities |
|
Identify significant impurities
Determine origin of impurities and method for elimination or reduction
Establish a control system for impurities involving: 1) Processing/manufacturing conditions 2)Suitable analytical methods/ specifications
|
Identify potential degradation product through stress testing and actual degradation products through stability studies.
Understand degradation pathway and methods to minimize degradation.
Establish a control system for impurities involving: 1) Processing/manufacturing conditions 2) Suitable analytical methods/ specifications 3) Long term storage conditions including packaging 4) Formulation. |
Following are the few examples of impurities which are reported in the API’S.
Table 2 Various impurities reported in API’s
|
Drug |
Impurity |
Method |
Reference |
|
AmphotericinB Atropine sulphate Cloxacillin Dextrose Doxorubicin Hydrochloride Ethambutol Hydrochloride Fluorescene sodium Framycetin sulphate
Mercaptopurine
Cimetidine
Norgestrel
Celecoxib
Ethynodiol diacetate
Methamphetamine
Repaglinide
Morphine Morphine sulphate
|
Tetraenes Apo atropine N,N dimethyl aniline 5 hydroxyl methyl furfural Acetone and ethanol
2 amino butanol
Dimethyl formamide Neamine
Hypoxanthine 2,5-bis[(N’-cyano-N’’-methyl) guinidinoethylthiomethyl]-4- methylimidazole and 1,8- bis[(N’ cyano- N’’- methyl)guinidino]-3,6 dithiaoctane 3,17α-diethinyl-13-ethyl 3,5- gonadiene-17-ol [5-(4-methylphenyl)-3 trifluromethyl-1H pyrazole], 4- [5-(2’-methylphenyl)-3- (trifluoromethyl)-1H pyrazol- 1-yl] benzenesulphonamide, and 4-[4-(4’-methylphenyl)-3 (trifluromethyl)-1H-pyrazole- 1-yl]-banzenesulfonamide 17 α-ethinyl-estr-4-ene-3β,17-diol-3-acetate-17-(3’-acetoxy- 2’-butenoate), 17 α-ethinylestr-4-ene-3β,17-diol-3- acetate-17-(3-oxo-butanoate) 1,2-dimethyl-3 phenylaziridine, ephedrine, methylephedrine, Nformylmethamphetamine, Nacetylmethamphetamine, Nformylphedrine, Nacetylephedrine, N,Odiacetylephedrine, methametamine dimmer 4-carboxymethyl-2-ethoxy benzoic acid, 4- cyclohexylaminocarbamoylmethyl- 2-ethoxy-benzoic acid, 1-cyclohexyl-3-[3-methyl-1-2- (piperidin-1-yl-phenyl)-butyl]- urea, 1,3-dicyclohexyl urea 6-monoacetylmorphine 5-(hydroxymethyl)-2-furfural, 10-hydroxymorphine, 10- oxomorphine
|
UV spectroscopy UV spectroscopy GC UV spectroscopy GC
TLC
GC TLC
UV spectroscopy
HPLC
TLC, HPLC and UV spectroscopy HPLC, LC, LC-MS-MS
HPLC
GC
HPLC
HPLC HPLC |
[24] [24] [24] [25] [25]
[25]
[25] [25]
[26]
[27]
[28]
[29]
[30]
[31]
[32]
[33] [33] |
9. Conclusion
Nowadays, it is mandatory requirements in various pharmacopoeias to know the impurities present in API’s. Isolation and characterization of impurities is required for acquiring and evaluating data that establishes biological safety which reveals the need and scope of impurity profiling of drugs in pharmaceutical research. To isolate and quantify the impurities, various instrumental analytical techniques are routinely been used.
10. Acknoeledgements
The author is thankful to Mr. Amardeep Ankalgi, Head of Pharmaceutical Chemistry department, B.N.College of Pharmacy, Udaipur for providing necessary data to carry out review work.
11. References
1.Gorog S, Identification and Determination of Impurities In Drugs, Elsevier Science Publishing Company, Amsterdam, published: May-2000, 154.
2.Sanjay B. Bari, Bharati R. Kadam, Yogini S. Jaiswal, Atul A. Shirkhedkar, Impurity profile: Significance In Active Pharmaceutical Ingredient, Eurasian Journal of Analytical Chemistry, Volume 2, Number 1, 2007, ISSN:1306-3057
3.Renu Solanki, Impurity Profiling Of Active Pharmaceutical Ingredient And Finished Drug Product, International Journal Of Drug Research And Technology,2012,vol-2(3),231-238,ISSN-2277-1506
4.International Conference on Harmonization, Draft Revised Guidance on Impurities in New Drug Substances. Federal Register Q3A(R), 2000, 65 (140): 45085.
5.ICH guideline, Q3Avalidation of analytical procedure: Methodology, 1996,6,11.
6.Michael E; Ira S. Krull; Analytical method development and validation, 1992,269.
7.United National drug control program, V.01-83778-May2001, P.No-65.
8.United State Pharmacopeia, The National Formulary, Asian Edition. 2004.
9.International Conference on Harmonization, Draft Revised Guidance on Impurities in New Drug Products. Federal Register Q3B(R), 2000, 65 (139): 44791.
10.International Conference on Harmonization, Impurities, Q3C- Guidelines for Residual Solvents, Q3C. Federal Register, 1997, 62(247): 67377.
11.International Conference on Harmonization, Specifications, Q6A: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products. Chemical substances, 1999, 65 (146):67488.
12.Yogesh Thorat, Significance of Impurity Profiling: A review, International Journal of Drug formulation & Research, Vol-2, Issue-4, Jul-Aug2011, 186.
13.Dorottya Barots,Sandor gorog,Recent Advances In Impurity Profiling Of Drug, Current Pharmaceutical Analysis,Vol-4,Issue-4,P.No:215-230(16),ISSN-1573-4129.
14.United States Pharmacopoeia XXIV, United States Pharmacopoeial Convention, INC, Rockville; 2000.
15.Talluri N K , Impurity Profiling of Drugs and Pharmaceuticals, New Analytical Methods- Development and Validation, LAP Lambert Academic Publishing, 2011, 14
16.Keval D. Parmar, Dr.N.M.Patel, Dr.P.M.Patel A Review: Impurity Profile Study, Journal of Pharmacy Research, Vol 4, No 8 (2011).
17.Mr.Anand M. Kudal, impurity profile of active pharmaceutical ingredient: A review, latest reviews, vol. 4 issue 5, pharmainfo.Net, pharmaceutical information, articles and blogs, 2006.
18.Alsante K M, Hatajik T D, Lohr L L and Sharp T R, Isolation and Identification of Process Related Impurities and Degradation Products from Pharmaceutical Drug Candidates. Part 1, American Pharmaceutical Review, 2001, 4(1): 70.
19.Peter J S; Ahmed A; Yan W, Journal of Pharmaceutical Biomedical Analysis, 2006, 41, 883.
20.Radhakrishna T, Satynarayana J and Satynarayana A, Determination of Loratidine and its Related Impurities by HPLC. Indian Drugs, 2002, 39 (6): 342.
21. Radhakrishna T, Satynarayana J and Satynarayana A (2002) HPLC method for the Degradation of Celecoxib and its Related Impurities. Indian Drugs, 2002, 40(3): 166.
22.Zawilla N H; Li B; Hoogmartens J; Adams E, Journal of Pharmaceutical Biomedical Analysis, 2006, 42, 114.
23.Lohr L L; Sharp T R; Alsante K M ; Hatajik T D ,American Pharmaceutical Review, 2001, 4(1), 130-139.
24.British Pharmacopoeia, the Department of Health, Social Services and Public Safety, 2004.
25.Indian Pharmacopoeia, Government of India, Ministry of Health and Family Welfare. Published by the Controller of Publications, 2004.
26.Halmos Z, Szantay C, Brlik J J, Csehi A, Varga K, Horvath P, Kislaki M, Domani G, Nemes A and Gorog S, Estimation of impurity profile of drugs and related materials Part 15. Identification of minor impurities in cimetidine. J Pharm Biomed Anal, 1996; 15: 1.
27.Horvath P, Balogh G, Brlik J, Csehi A, Dravecz F, Halmos Z, Lauko A, Renyei M, Varga K and Gorog S, Estimation of impurity profile of drugs and related materials Part 16: identification of the side-products of the ethinylation step in the synthesis of contraceptive gestogens. J Pharm Biomed Anal, 1997; 15:1343
28.Satyanarayana U, Sreenivas Rao D, Ravindra Kumar Y, Moses Babu J, Rajender Kumar P and Tirupathi Reddy J, Isolation, synthesis and characterization of mpurities in celecoxib a COX-2 inhibitor. J Pharm Biomed Anal, 2004; 35: 951.
29.Babjak M, Balogh G, Gazdag M and Gorog S, Estimation of impurity profile of drugs and related materials Part XXI. HPLC/UV/MS study of the impurity profile of ethynodiol diacetate. J Pharm Biomed Anal, 2002; 29:1153
30.Vichet P, Narini S, Juthamard P, Wiphada P, Tetsuro S and Ken T, Idetification of impurities and statistical classification of methamphetamine tablets (Ya-Ba) seized in Thailand. Forensic Science International, 2002; 126:105.
31.Krishna Reddy K V S R, Moses Babu J, Vijayvitthal T M, Eswaraiah S, Satyanarayana Reddy M, Dubey P K and Vyas K, Impurity profile study of repaglinide. J Pharm Biomed Anal, 2003; 32:461
32.Dams R, Benijts T, Lambert W, Massart D and De Leenheer A, Heroin impurity profiling: trends throughout a decade of experimenting-Review. Forensic Science International 2001; 121:81
33.Kelly S, Padraig M G, Stuart J M and David H G , An impurity in a morphine sulphate drug product identified is 5-(Hydroxymethyl)-2-furfural, 10- Hydroxymorphine and 10-Oxomorphine. J Pharm Biomed Anal 2003; 92:485
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








