
 About Authors:
About Authors:
Sahithya Bitragunta, Ponnuru Venkata Suresh, Nadendla RamaRao
Chalapathi institute of pharmaceutical sciences,
Lam,Guntur,
India.
Abstract:
In 1960’s, different countries used to have different set of guidelines and laws for the registration and marketing of pharmaceuticals. But tragedies like Thalidomide disaster made harmonization of all technical requirements which are required for the registration of pharmaceuticals for the human use. Thus ICH has been launched in 1990 by drug regulatory authorities and pharmaceutical industrial members of Europe, Japan and US with an objective of providing guidelines to ensure the registration and marketing of safe, effective and high quality medicines.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1205
Introduction:
ICH is the “International conference on Harmonization of technical requirements for the Registration of Pharmaceuticals for human use.
To assure safety, quality and efficacy of medicines, the members of ICH who include members from drug regulatory authorities and research based industries of European Union, US and Japan will discuss on the technical procedures and documents required.
History behind the initiation of ICH:
Thalidomide disaster:
In 1960’s Thalidomide has been approved for marketing in Europe and Africa for treating morning sickness in pregnant women’s. But during approval for marketing in US, the drug has been rejected by a Canadian lady DR. Frances O.Kelsey, a Canadian pharmacologist who was working as a reviewer of Thalidomide in US Food and Drug administration stating that the drug has serious neurological effects.
But before getting approval the drug has been distributed freely as physician samples in US this has lead to the delivery of limulus babies.
Lack of proper policies, Legislations and guidelines to ensure the safety, efficacy of the drug made 10,000 to 20,000 babies suffer from birth defects like phocomelia.
This disaster has initiated many policies, Legislations which has to be brought to ensure the safety, efficacy, quality of the drugs, thus leading to International conference on harmonization in 1990.
Need of Harmonization:
The difference in the technical requirements and the procedures followed by different countries made the global marketing of drug time consuming and expensive.
So, to reduce the cost and time required for the global marketing of a drug , harmonization of technical requirements has been promoted and special guidelines has been framed to ensure the quality, safety and the efficacy of the drug.
Objective of ICH:
The main objective of ICH is to
* Promote international harmonization of technical requirements to develop safe, effective, and high quality medicines.
* Reduce the registration cost.
* Promote public health.
* Prevent the duplication of clinical trialsin Humans
* Minimize the animal use without compromising in the safety, qualitut compromising in the safety, quality.
Goal of ICH:
The goal of ICH is to
* Promote international harmonization by bringing together representatives from the three ICH regions (EU, Japan and USA) to discuss and establish common guidelines.
* Make information available on ICH, ICH activities and ICH guidelines to any country.
Members of ICH:
• ICH is comprised of representatives from six parties that represent the regulatory bodies and research based industry in the European Union, Japan and the USA.
• In Japan, the members are the Ministry of Health, Labour and Welfare (MHLW), and the Japan Pharmaceutical Manufacturers Association (JPMA). In Europe, the members are the European Union (EU), and the European Federation of Pharmaceutical Industries and Associations (EFPIA).
• In the USA, the members are the Food and Drug Administration (FDA), and the Pharmaceutical Research and Manufacturers of America (PhRMA).
• Additional members include Observers from the World Health Organization (WHO), European Free Trade Association (EFTA), Canada.
Organization of ICH:
The organizational structure of ICH consists of the following components:
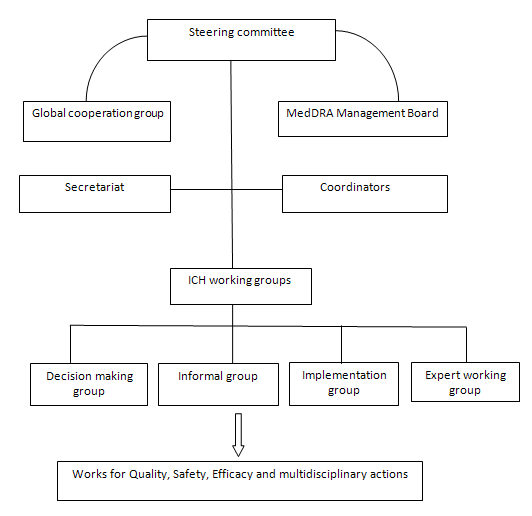
ICH steering committee:
Responsibilities:
The ICH Steering Committee (SC) is the main organization which is involved in the following works
• Governs ICH
• Determines the policies and procedures for ICH.
• Selects topics for hormonisation
• Monitors the progress of harmonization initiatives
Members:
ICH is comprised of representatives from the regulatory bodies and research -based industry in the European Union, Japan and the USA.
Voting members:
Members from Japan:
• Ministry of Health, Labour and Welfare (MHLW)
• Japan Pharmaceutical Manufacturers Association (JPMA).
Members from Europe :
• European Union (EU)
• European Federation of Pharmaceutical Industries and Associations (EFPIA).
Members from USA:
• Food and Drug Administration (FDA)
• Pharmaceutical Research and Manufacturers
Non-Voting members or ICH Observers:
• World Health Organization (WHO)
• Health Canada and the European Free Trade Association (EFTA)
• IFPMA
IFPMA hosts the ICH Secretariat and participates as a non-voting member of the SC
ICH coordinators:
The Coordinators are fundamental to the smooth running of the ICH and are nominated by each of the six parties of ICH. Coordinator acts as the main contact point with the ICH Secretariat.
Secretariat:
Location:
• Operates from the IFPMA offices in Geneva, Switzerland
Responsibilities:
• provides support to the ICH Steering Committee.
• Documents the meetings of the steering committee.
• Promotes coordination between Working Groups.
• Provides information on the ICH guidelines and ICH process.
• Provides administrative support for MedDRA management board.
• Provides administrative support for Global cooperation Group.
[adsense:468x15:2204050025]
ICH Global cooperation Group(GCG):
ICH Global cooperation Group has been formed onMarch 11, 1999, as a subcommittee of ICH steering committee to Globalise the ICH and its guidelines.
Members:
Members from ICH:
One representative from each of the six parties of ICH steering committee
ICH secretariat at IFPMA
ICH Observers:
• World Health Organization (WHO)
• Health Canada and the European Free Trade Association (EFTA)
• IFPMA
Non-ICH Members: They include
Regional Harmonisation initiatives(RHIs)
• Asia-pacific Economic Cooperation(APEC)
• Association of Southeast Asian Nations(ASEAN)
• Gulf Cooperation Council(GCC)
• Pan American Network on Drug RegulatoryHormonization (PANDRH)
• South African DevelopmentCommunity(SADC)
RHIs are involved in hormonising the drug regulation across a defined group of Non-ICH countries.
• Drug Regulatory Authorities and Department of Health of Australia, Brazil,China, India, Russia, Singapore, South Korea forms a permanent representative of GCG.
Working groups:
• Each party establishes a Contact Network of experts within their own organisation or region in order to ensure that, in the discussions, they reflect the views and policies of the co-sponsor they represent.
• For each of the technical topics which have been selected for harmonisation in the first phase of activities, the SC appointed a Working Group to review the differences in requirements between the three regions and develop scientific consensus required to reconcile those differences.
• Working groups do not have a fixed "membership" but each of the six parties have nominated a Topic Leader (and, frequently, a Deputy Topic Leader) as the contact for the topic.
• The Observers to ICH, the Pharmacopoeial authorities and representatives from the self-medication industry and the generic industry have been invited to participate in various working groups.
Depending on the type of harmonisation activity needed, the working groups are classified as follows by the Steering Committee.
Expert Working Group:
It is created by the Steering Committee when a new topic is accepted for harmonisation, and is charged with developing a harmonised guideline that meets the objectives outlined in the Concept Paper and Business Plan.
Implementation Working Group :
It is established by the Steering Committee to develop Q&As to facilitate the implementation of existing guidelines.
Informal Working Group :
It is either an informal EWG or IWG formed prior to any official ICH harmonisation activity with the objectives of developing/finalising a Concept Paper, as well as developing a Business Plan.
Discussion group:
It is a group established to discuss specific scientific considerations or views.For example Gene therapy discussion group, ICH and women discussion group.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Steps in the ICH:
Step 1:
Drafts are prepared and circulated through many revisions until a "final harmonised draft" is completed under the leadership of the Rapporteur.
Step 2:
This draft is signed by the Expert working group(EWG) as the agreed-upon draft and forwarded to the Steering Committee for signing which signifies acceptance for consultation by each of the six co-sponsors
Step 3:
The three regulatory sponsors initiate their normal consultation process to receive comments . This comment period normally takes six months. The draft document to be generated as a result of the Step 3 phase is called Step 4 Experts Document.
If both regulatory and industry parties of the EWG are satisfied that the consensus achieved at Step 2 is not substantially altered as a result of the consultation, or consensus is reached on any alterations, theStep 4 Experts Document is signed by the EWG regulatory experts. The Step 4 document with regulatory EWG signatures is submitted to the Steering Committee to request adoption as Step 4 of the ICH process.
Step 4:
It is reached when the Steering Committee agrees that there is sufficient scientific consensus on the technical issues. This endorsement is based on the signatures from the three regulatory parties to ICH affirming that the Guideline is recommended for adoption by the regulatory bodies of the three regions.
Step 5
The process is complete when the guidelines are incorporated into national or regional internal procedures
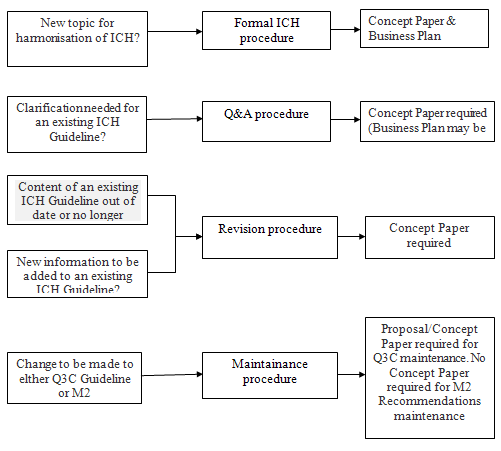
Process of harmonization:
ICH harmonization falls into four categories Formal ICH procedures, Revision procedure, Maintenance procedure.
MedDRA Management Board :
MedDRA:
MedDRA is a highly standardised medical dictionary developed by ICH to facilitate the sharing of regulatory information internationally for medical products used by humans.
It is used for the registration, documentation and safety monitoring of medical products before and after marketing.
MedDRA products:
The products include pharmaceuticals, vaccines and drug-device combination products.
MedDRA users:
The utilization of MedDRA is increasing day by day and the current users of MedDRA include
Regulatory authorities
Global pharmaceutical companies
Clinical research organisations
Health care professionals
MedDRA provides terminology which is used globally during reporting all the phases of a drug development.
MedDRA management board:
It is appointed by ICH Steering Committee .
Members:
One representative from each of the six ICH parties.
One representative from Medicines and Healthcare products Regulatory agency of UK and Health Canada
IFPMA acts as a non-voting member .
Responsibilities:
Oversee the activities of MedDRA, MSSO ( maintainance and support services organisation)
Ensure that MSSO is meeting the various needs of MedDRA users Access:
MedDRA is available on subscription from the MedDRA Maintainance and suport organisation
IT is available freely for Healthcare providers, regulators and non-profit organisors.
ICH guidelines:
The ICH topics are divided into four categories and ICH topic codes has been given according to these categories.
1. Quality guidelines.
2. Safety guidelines.
3. Efficacy guidelines.
4. Multidisciplinary guidelines.
Q-Quality guidelines
It provides guidelines regarding the Harmonization achievement in quality area, conduct, stability studies, defining relevant thresholds for impurities testing & more flexible approach to pharmaceutical quality based on (GMP) risk management
S-Safety guidelines
It provides safety guidelines to uncover potential risks like carcinogenicity, genotoxicity and reprotoxicity.
E-Efficacy guidelines
It is concerned with design, conduct, saftey and reporting of clinical trials; it also covers novel types of medicines derived from biotechnological process and the use of pharmacogenitic/genomics techniques to produce better targeted medicines
M-Multidisciplinary guidelines
Includes ICH medical terminology and common technical document and the development of electronic standards for the transfer of regulatory information.
Q-Quality guidelines:
|
Q1A-Q1F |
Stability studies |
|
Q1A |
Stability testing of new drug substances & products |
|
Q1B |
Photo stability testing of new drug substances & products |
|
Q1C |
Stability testing of new dosage forms |
|
Q1D |
Bracketing & matrixing designs for stability testing of new drug substances & products |
|
Q1E |
Evaluation of stability data |
|
Q2 |
Validation of analytical procedures |
|
Q3A-Q3D |
impurities |
|
Q3C,Q3D |
Guidelines for residual solvents, metal impurties |
|
Q4-Q4B |
Pharmacopoeias |
|
Q5-Q5E |
Quality of biotechnology products |
|
Q6A-Q6B |
Specifications for new drug substances & products |
|
Q7 |
Good manufacturing practice |
|
Q8 |
Pharmaceutical development |
|
Q9 |
Quality risk management |
|
Q10 |
Pharmaceutical Quality system |
Safety guidelines
|
S1A |
Guideline on the need for carcinogenicity studies of pharmaceuticals |
|
S1b |
Testing for the carcinogenicity of pharmaceuticals |
|
S1C |
Dose selection for carcinogenicity |
|
S2A |
Guidance on specific aspects of regulatory genotoxicity tests for pharmaceuticals |
|
S2B |
Genotxicity |
|
S2C |
Guidance for Genotoxicity testing and data interpretation |
|
S3A |
Guidance on toxicokinetics, the assessment of systemic exposure in toxicity studies |
|
S3B |
Pharmacokinetics:Guidance for repeated dose tissue distribution studies. |
|
S4 |
Duration of chronic toxicity testing in animals |
|
S5 |
Detection of toxicity to reproduction for medical products and toxicity to male fertility |
|
S6 |
Perclinical safety evaluation of biotechnology-derived pharmaceuticals |
|
S7 |
Pharmacology studies |
|
S7A |
Non-clinical evaluation of the potential for delayed ventricular repolarisation by human pharmaceuticals. |
|
S8 |
Immunotoxicity studies |
|
S9 |
Non-clinical evaluation for Anticancer drugs. |
Efficacy guidelines
|
E |
Clinical safety for drugs intended for long term treatment of non-life threatening conditions. |
|
E2A |
Clinical safety data management |
|
E2B(R5) |
Implementation working group questions & answers |
|
E2B(R2) |
Maintenance of ICH guidline on Clinical safety |
|
E2B(R1) |
Clinical safety & periodic safety for marketed drugs |
|
E2D |
Post-approval safety data management |
|
E2E |
Pharmacovigilance planning |
|
E2F |
Development safety update report |
|
E3 |
Structure and content of clinical study reports |
|
E4 |
Dose-response information to support drug registration |
|
E5 |
Implementation working group questions & answers |
|
E5(R1) |
Ethnic factors in the acceptability of foreign clinical data |
|
E6 E6(R1) |
Guidline for good clinical practice |
|
E7 |
Studies in support of special populations: questions & answers |
|
E8 |
Staistical principles for clinical trails |
|
E9 |
Statistical principles for clinical trials |
|
E10 |
Choice of control group & related issues in clinical trails |
|
E11 |
Clinical investigation of medicinal products in the pediatric population |
|
E12 |
Clinical evaluation of new anti-hypertensive drugs |
|
E14 |
Clinical evaluation for non- antiarrhytmic potential |
|
E15 |
Biomarkers related to drug for biotechnology product development |
|
E16 |
Biomarkers related to drug for biotechnology product development |
Multidisciplinary guidelines
|
M2 |
Electronic transmission of individual case safety reports message specification |
|
M3 |
Guidance on non-clinical safety studies for the conduct of human clinical trails & marketing authorization for pharmaceuticals |
|
M4 |
Organization of the common technical document for the registration of pharmaceutical for human use |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
The Impact of ICH (Efficacy) on Industry
• Repeat clinical trials were a fact of life in drug development if a company wished to market a drug in more than one region. This costly and time consuming activity, frequently involving the repeat of long, resource intensive Phase III clinical trials, is obviated in most cases by the introduction of Ethnic Factors in the Acceptability of Foreign Clinical Data” (E5) guideline.
• When the trials are run in accordance with the principles of GCP laid down in the ICH guideline “Good Clinical Practice: Consolidated Guideline” (E6), foreign clinical trial data may be submitted in support of a submission in any ICH region.
• Less than a year after the guideline was finalized Pfizer was able to apply it to great effect to gain approval of Viagra® in Japan by use of a bridging study (a key part of the E5 guideline), rather than a repeated clinical trial(s) as would have been required previously.
• The guideline “General Considerations for Clinical Trials” (E8), provides a set of internationally accepted principles to be applied to trial design, which aids the acceptance of data throughout the three regions.
• The guideline E2A (Clinical Safety Data Management: Definitions and Standards for Expedited Reporting) and E3 (Structure and Content of Clinical Study Reports). E2A led to a harmonization of expedited reporting in the three regions, defining when clinical study reports are required and the amount of detail they should contain. It also ensures that reporting times are measured in calendar days rather than working days .
• E3 (Clinical Study Reports), like E6 (GCP), early ICH process, establishing a common format for clinical study reports. This common presentation has made preparing multiple regulatory submissions a far simpler process as a single core document (with appendices) may be used in all three ICH regions.
• One of the most important outcomes of the harmonization work in the efficacy area, as well as the recognized reduction in time and resources used in a development program, is that the unified operating practices enhanced patient safety in the clinical trials process
The Impact of ICH (Quality) on Industry
• The ICH guidelines in the Quality area have provided recommendations in two of the key areas that define bulk drug and drug product quality—stability data and impurities—and led to a significant reduction in duplicate testing.
• Prior to these guidelines there was no harmonized approach to the data requirements in these areas. With stability for example, it was typical to run studies at “room temperature” as defined by the company concerned, and appropriate to the locality.
• There was also no humidity control. This resulted in registrations in different regions requiring new stability data if the climatic zone was different to that where the original study had been conducted.
• ICH harmonization provided standard sets of conditions taking account of the climatic zones in each of the three regions.
• This means that the information on stability generated in any one of the three regions is mutually acceptable in the other two areas, provided it meets the requirements of the guideline. This removed the duplicate testing .
• The impurities guidelines (Impurities in New Drug Substances (Q3A), Impurities in New Drug Products (Q3B), and Impurities: Guideline for Residual Solvents (Q3C)) also served, as with the stability guidelines, to provide scientific agreement on the recording and reporting of impurity levels
• Guidelines were also provided on how changes in impurity profile over the course of a development program should be managed. The result of this is that it should be possible to determine a single specification for any drug substance or product that is acceptable across the three ICH regions. This makes the supply chain far simpler, and minimizes supply error.
• ICH has also produced a parallel set of guidelines covering the specific issues associated with biotechnological products. Standardization through the guidelines has been a very positive step for the biotechnology industry, and has certainly had a significant favorable impact on both development times and resource utilization.
• Duplication of research was reduced related to the stability testing, impurity profiles.
The Impact of ICH (Safety) on Industry
• The guidelines in this area have very much represented industry’s current best practice.
• By a careful examination of standard practice and the types of data that could be accessed from studies the EWGs in this area were able to determine what testing was necessary to examine any one type of toxicity, and thus to generate a standard battery of tests.
• This resulted in the guidelines comprehensively covering carcinogenicity testing, genotoxicity testing, reprotoxicity testing, chronic toxicity testing.
• There is also a guideline concerning biotechnology derived pharmaceuticals, and guideline for the Timing of Non-Clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals (M3). This defined the safety data that must be available before human volunteers or patients may be treated with the new drug.
• There is a standard set of tests recommended for most types of toxicity studies; timing, exact requirements (including dose) and need for toxicity studies for different indications or treatment durations have been defined.
• For carcinogenicity studies only one long-term study (usually carried out in a rodent species) plus one short- or mid-term study is needed. The safety guidelines made long term studies easy.
• currently, with the results available at the end of 2000); the special case of biotechnological products has also been considered. All of these resulted in a reduction in duplicate testing.
• As safety testing is an area of considerable research effort, both in academia and industry, an important result of, for example, the reduction in the number of long term studies that is required should be that (as well as reducing the use of animals) it will allow more resource to be diverted to other approaches to uncover potential risks like genotoxicity and carcinogenicity relevant to humans.
• The continual development of the models used to study toxicity is key to the industry becoming better able to evaluate the safety of new drugs, and delivering safe therapies to patients.
• It is on the basis of such research developments that ICH tries to keep guidelines updated and under review.
Conclusion:
ICH, through its activities in the harmonization of regulatory requirements across the EU, Japan and US, is enabling industry to reduce development times by removing the duplication of studies that was previously required to gain market approval for a new drug in each of the three regions. Industry has three reasons to support ICH and its continued efforts to further harmonize the technical requirements for the registration of innovative drugs which are reduced development times and resources, including an end to duplicate clinical trials due to ethnicity differences, easier simultaneous launch of a new drug in many countries (including across the three ICH regions), ICH guidelines as a recognized standard , will facilitate intra-company Globalization. Harmonization through ICH brings important, life-saving treatments to patients faster.
References:
1. ich.org
2. ich.org/about/organisation-of-ich.html
3. ich.org/about/organization-of-ich/steering.html
4.ich.org/about/organization-of-ich/working-groups.html
5. ich.org/products/meddra.html
6. ich.org products/guidelines.html
7. ich.org/about/faqs.html
8. ich.org/fileadmin/public-webbsite/ICH-products/Guidelines/quality
9. ich.org/about/process-of-hormonisation.html
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








