
About Authors:
Najneen S. Shaikh1*, Mr. MAYURESH. K. RAUT2
1Sinhgad Institute of Pharmaceutical Sciences (SIPS), Lonavala
2Asst. Professor Of Phamaceutical Chemistry
Sinhgad Technical Education Society,
Kusgaon (Bk), Lonavala- 410401
*naj.pharma.23@gmail.com
ABTRACT
Glycogen synthase kinase 3 (GSK-3) is a serine/threonine protein kinase, that mediates the addition of phosphate molecules on certain serine and threonine amino acids in particular cellular substrates.
Glycogen synthase kinase-3 (GSK3) has recently been linked to mood disorders and schizophrenia, and the neurotransmitter systems and therapeutic treatments associated with these diseases. GSK3 is a widely influential enzyme that is capable of phosphorylating, and thereby regulating, over forty known substrates. Four mechanisms regulating GSK3 (phosphorylation, protein complexes, localization, and substrate phosphorylation) combine to provide substrate-specific regulation of the actions of GSK3. Several intracellular signaling cascades converge on GSK3 to modulate its activity, and several neurotransmitter systems also regulate GSK3, including serotonergic, dopaminergic, cholinergic, and glutamatergic systems. Because of changes in these neurotransmitter systems and the actions of therapeutic drugs, GSK3 has been linked to the mood disorders, bipolar disorder and depression, and to schizophrenia.
[adsense:336x280:8701650588]
Inhibition of GSK3 may be an important therapeutic target of mood stabilizers, and regulation of GSK3 may be involved in the therapeutic effects of other drugs used in psychiatry. Dysregulated GSK3 in bipolar disorder, depression, and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression, and the ability of neurons to respond to stressful, potentially lethal, conditions. In part because of these key actions of GSK3 and its associations with mood disorders and schizophrenia, much res Glycogen synthase kinase 3 regulates glycogen synthase, the rate-determining enzyme for glycogen synthesis. GSK is enzyme which bind to the different receptor in CNS like NMDA ,Benzodizepam and GABA nergic Receptor,and its stimulation causes abnormal disorders in CNS like schizophrenia,bipolar disorders,mood disorders, Alziemer disorders, etc. so we need to inhibit these enzyme in CNS by some inhibitor which use in treating CNS disorders.
Reference Id: PHARMATUTOR-ART-1410
INTRODUCTION
Glycogen synthase kinase 3 (GSK-3) is a serine/threonine protein kinase, that mediates the addition of phosphate molecules on certain serine and threonine amino acids in particular cellular substrates. [1,2,3]
CNS DISORDERS: Alziemer disease,Bipolar disorders,schizophrenia,Mood disorder,Parkinson’s desease.
Types of glycogen synthase kinase
1. Glycogen Synthase Kinase alfa.
2. Glycogen Synthase Kinase beta.
GSK STRUCTURE
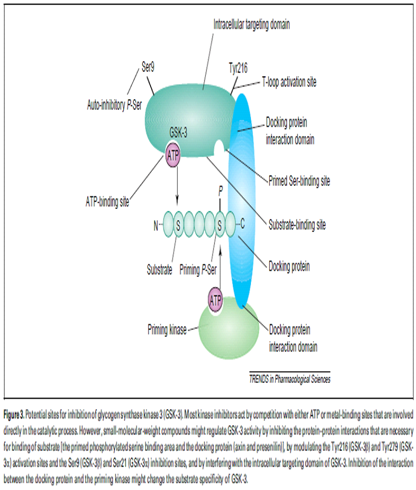
The overall shape is shared by all kinases , with a small N-terminal lobe, which consists mostly of b-sheets and a large C-terminal lobe, which is formed essentially of a-helices . The ATP-binding pocket is located between the two lobes.Arg96, Arg180 and Lys205 form a small pocket where the phosphate group of the primed substrate and the pseudo substrate bind GSK-3 is regulated at multiple levels.
First,GSK-3b is regulated by post translational phosphorylation of Ser9 (inhibitory) and Tyr216 (activating) (Ser21and Tyr279, respectively, in GSK-3a). Unphosphorylated Tyr216 in the T-loop domain prevents access of substrates to the catalytic site, and phosphorylation releases this inhibition.
Second,GSK-3b is regulated by interactions with many other proteins. Axin and presenilin act as docking proteins that allow the substrates to make contact with the priming kinase [casein kinase (CK1) and protein kinase A, respectively] and GSK-3 Third, GSK-3action requires the priming phosphorylation of its substrates by another kinase on a serine residue located four amino acids C-terminal to the GSK-3 phosphorylation site. Fourth, GSK-3 is regulated through its intracellular distribution.
Pharmacological inhibitors of glycogen synthase kinase3
GSK-3, as part of a multi-protein complex that contains proteins such as axin, presenilin and b-catenin, contains many additional targetsites for specific modulation activity. Protein phosphorylation, the most common post-translational mechanism used by cells to regulate enzymes 4,5

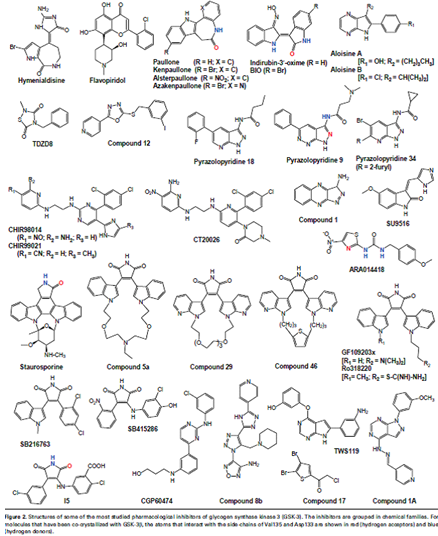
Pharmacological inhibitors mechanism of action
GSK-3-dependent abnormal phosphorylation of Tau and production of amyloid-b in Alzheimer’s disease have stimulated the search for potent, selective inhibitors of GSK-3. after the crystallization of GSK-3b, the enzyme was cocrystallized with some inhibitors, which provided an exquisite understanding of their mechanism of interaction within the ATP-binding pocket.Despite wide chemical diversity, most pharmacological inhibitors of GSK-3 share common properties:
• they have a low molecular weight(!600);
• they are rather flat, hydrophobic heterocycles;
• most, but not all, act by competing with ATP in the ATP-binding site of the kinase (lithium acts directly by competing with magnesium, but also indirectly by increasing the inhibitory phosphorylation of Ser9 and Ser21 of GSK-3b and GSK-3a, respectively ;
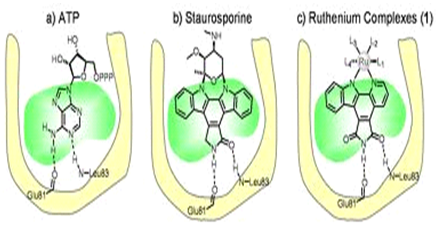
• like inhibitors of cyclin-dependent kinases (CDKs), they essentially bind through hydrophobic interactions and 2–3 hydrogen bonds with the kinase; and
• the backbone carbonyl and amino side-chains of Val135 act as an H-bond acceptor and H-bond donor, respectively, to the inhibitors, whereas the backbone carbonyl of Asp133 often acts as an H-bond acceptor .These interactions are similar to those described for CDK inhibitors ,. In general, compounds that interfere with the intracellular localizationof GSKmight constitute alternative ways to modulate the physiological functions of GSK.(7)
The selectivity of inhibitors
Because theATP-binding pockets ofGSK-a andGSK-b are similar, inhibitors that target these sites are unlikely to differentiate between the two isoforms. Such selectivity might only be achieved by drugs that act at other sites on the kinases, The selectivity of most of the available. Lithium has alternative targets such as inositol-phosphate phosphatases. Paullones,especially kenpaullone 6-bromo-substituted indirubinsand the azaindolylmaleimide compound 46 , appear to be among the most selective inhibitors identified so far.
GSK inhibitors: diversity of applications
Nervous system disorders
The beneficial effect of lithium in the treatment of bipolar affective disorder might result from GSK inhibition but lithium use alternatively because of it side effect. Due to Alzheimer’s disease (AD) is characterized by three essential events, mutation of any one of the genes that encode presenilin-1, presenilin-2and the b-amyloid precursor protein (b-APP) results in w100% penetrance;
GSK a is required for the production of amyloid-b] and amyloid-b toxicity is mediated by increased GSK-3 activity.
Last, GSK-3, with CDK5 and microtubule affinity-regulating kinase (MARK), is one of the major kinases that is involved in abnormal Tau phosphorylation on AD-specific sites. Presenilin-1 interacts directly with GSK-, which favors the interaction of GSK with substrates such as Tau. Together, these data indicate the potential benefits of GSK-3 inhibition in counteracting the three major AD events, presenilin action, amyloid-b peptide production and Tau hyperphosphorylation.
GSK-3 is involved in neuronal cell death [10]. There is experimental evidence that reducing GSK-3 activity by small-interfering RNS (siRNA), lithium and kenpaullone in cell lines, and by lithium in mice, reduces amyloid-b production [11] . Together, these and other data strongly encourage the evaluation of GSK-3 inhibitors as neuroprotective agents in AD. Because CDK5 is also involved in neurodegeneration, and mitotic CDK1 and its upstream activators are expressed in the brains of AD patients but not of normal individuals ,. Lithium is reported to protect neuronal cells in culture from excite toxicity. Lithium reduces the neurological deficits and decreases the brain-infarct size when given before or after middle cerebral- artery occlusion in a Parkinson’s disease (PD) is a neurodegenerative disease. that is characterized by the specific loss of dopamine containing neurons. Sustained stimulation of dopamine receptors b leads to GSK-3 activation, and several GSK-3 inhibitors appear to partially compensate dopamine-dependent 6.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
REVIEW OF LITERATURE
Glycogen synthase kinase-3 (GSK3) has recently been linked to mood disorders and schizophrenia, and the neurotransmitter systems and therapeutic treatments associated with these diseases.
GSK3 is a widely influential enzyme that is capable of phosphorylating, and thereby regulating, over forty known substrates.
Four mechanisms regulating GSK3 (phosphorylation, protein complexes, localization, and substrate phosphorylation) combine to provide substrate-specific regulation of the actions of GSK3. Several intracellular signaling cascades converge on GSK3 to modulate its activity, and several neurotransmitter systems also regulate GSK3, including serotonergic, dopaminergic, cholinergic, and glutamatergic systems. Because of changes in these neurotransmitter systems and the actions of therapeutic drugs, GSK3 has been linked to the mood disorders, bipolar disorder and depression, and to schizophrenia. Inhibition of GSK3 may be an important therapeutic target of mood stabilizers, and regulation of GSK3 may be involved in the therapeutic effects of other drugs used in psychiatry.
Dysregulated GSK3 in bipolar disorder, depression, and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression, and the ability of neurons to respond to stressful, potentially lethal, conditions. In part because of these key actions of GSK3 and its associations with mood disorders and schizophrenia, much research is currently being devoted to identifying new selective inhibitors of GSK3.
The association of glycogen synthase kinase-3 (GSK3) with psychiatric diseases is a relatively new concept, first having been raised only in 1996. At that time, it was discovered that GSK3 is a target of the mood stabilizer lithium , a primary treatment for bipolar mood disorder ,an illness also referred to as manic-depression. During the intervening ten years, a wide variety of types of studies have contributed to the hypothesis that inhibition of GSK3 by lithium makes an important contribution to lithium’s mood stabilizing capability .
These recent findings not only support the proposition that inhibition of GSK3 is an important therapeutic target of mood stabilizers, but also indicate that regulation of GSK3 may be involved in the therapeutic effects of other drugs used in psychiatry. Thus, evidence has begun to accumulate suggesting that dysregulation of GSK3 may occur in depression and schizophrenia, as well as in bipolar disorder.
Equally important to substantiating the link between GSK3 and psychiatric diseases are recent studies of the functions of GSK3. These studies have provided strong mechanistic hypotheses concerning how neuronal plasticity and function could be impaired by abnormally regulated GSK3 in psychiatric diseases. These diverse findings linking GSK3 to psychiatric diseases are summarized and evaluated in this review.
REGULATION OF GSK
The major physiological mechanism that regulates the activity of GSK3 is phosphorylation of an N-terminal serine of GSK3 .This serine phosphorylation inhibits the activity of GSK3 and can be catalyzed by several kinases, such as Akt (Fig. 1). Thus, many growth factors that activate receptors coupled to the sequential activation of phosphoinositide 3-kinase (PI3K) and Akt inhibit GSK3 activity by increasing the Akt-mediated phosphorylation of the regulatory serine of GSK3. As shown in Fig. (1), other prominent intracellular signaling pathways, including those that activate protein kinase A or protein kinase C, also converge on GSK3 to inhibit it via phosphorylation of the N-terminal serine.
Regulation of the inhibitory serine-phosphorylation of GSK3
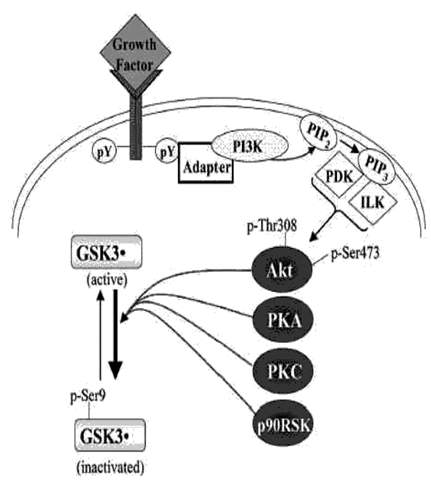
Regulation of the inhibitory serine-phosphorylation of GSk.GSK Phosphorylation of Ser-9 of GSK3β inhibits its activity.
Some of the kinases reported to phosphorylate this site on GSK3β include Akt, protein kinase A (PKA; also known as cyclic AMP-dependent protein kinase), protein kinase C (PKC), and p90 ribosomal S6 kinase (p90RSK).
Signaling leading from a growth factor receptor to activation of Akt is depicted. Growth factor receptor stimulation causes tyrosine phosphorylation (pY) of the receptor which interacts with various adaptor proteins to initiate a signaling cascade that results in activation of Akt via dual phosphorylation on threonine-308 and serine-473 which results in Akt-induced serine-phosphorylation and inactivation of GSK3β .
ILK, integrin-linked kinase; PDK, phosphoinositide-dependent kinase; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate.
Further, substrate-selective regulation of the actions of GSK3 is also needed because GSK3 phosphorylates more than 40 substrates. This large number of substrates enables GSK3 to influence many critical cellular functions, such as gene expression, cell structure, neural plasticity, and survival, so regulatory mechanisms must be invoked to selectively alter GSK3 activity to limit the substrates that it phosphorylates.
GSK3-binding proteins provide one method by which cells have developed substrate-selective regulation of GSK3 (Fig. 2A). For example, in the Wnt signaling pathway axin and other proteins bind GSK3 to direct its actions to a specific substrate, β -catenin.
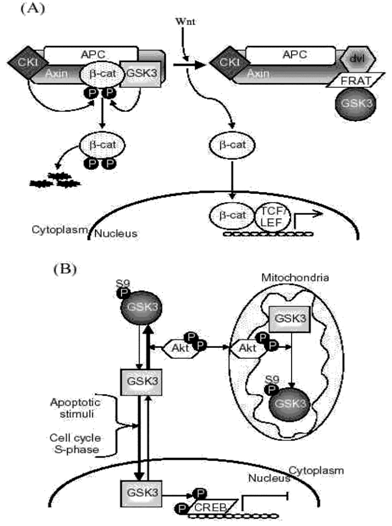
Fig2:Mechanisms contributing to substrate-selective regulation of GSK
Recently several additional GSK3-binding proteins have been identified and it appears that this is a common mechanism by which the action of GSK3 is directed to specific substrates
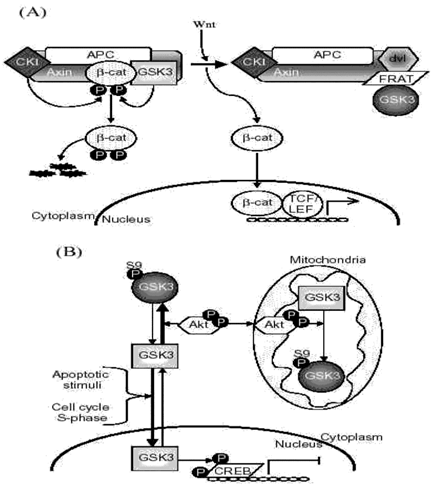
2:Mechanisms contribution
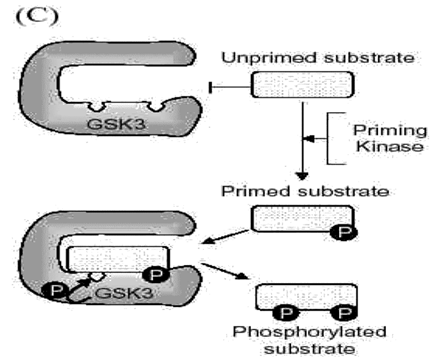
Fig 2: Mechanisms contributing to substrate-selective regulation of GSK3
A. GSK3-binding proteins. GSK3-binding proteins regulate the action of GSK3 in the Wnt signaling pathway. Axin acts as a scaffold bringing together the substrate β -catenin with APC, the priming kinase, casein kinase 1 (CK1), and GSK3. In the absence of Wnt, phosphorylation of β -catenin by CK1 and GSK3 targets it for degradation. Wnt activation results in disheveled (dvl) and FRAT binding to GSK3, inhibiting its phosphorylation of β -catenin. This results in the stabilization and accumulation of β -catenin, its translocation to the nucleus, and facilitation of TCF/LEF-mediated transcription.
APC, adenomatous polyposis coli gene product; FRAT, frequently rearranged in advanced T-cell lymphoma; LEF, lymphoid-enhancing factor; TCF, T cell factor.
B. Subcellular distribution. GSK3 is predominantly a cytosolic protein, where it can be regulated by serine phosphorylation carried out by several kinases, such as Akt. GSK3 reversibly translocates to the nucleus where it’s level is increased in the S-phase of the cell cycle and during some types of apoptosis. Nuclear accumulation of GSK3 facilitates its actions on nuclear substrates, such as the transcription factor cyclic AMP response element-binding protein (CREB), which is inhibited following phosphorylation by GSK3. Mitochondria also contain GSK3. Although no studies have reported alterations of mitochondrial GSK3 levels, its activity can be regulated by serine phosphorylation. For example, activation of cytosolic Akt can lead to its import into mitochondria where it can serine-phosphorylate mitochondrial GSK3 to inhibit its activity.
C. Priming phosphorylation of GSK3 substrates. Many substrates of GSK3 must be "primed", which means they are pre-phosphorylated at a serine/threonine four residues removed from the serine/threonine that is phosphorylated by GSK3. Thus, the consensus site for phosphorylation of primed substrates by GSK3 is S/T-X-X-X-S/T(p). This provides an important regulatory mechanism controlling the action of GSK3 because signaling pathways phosphorylating its substrates must be active before GSK3 can have any effect on such substrates. The preference of GSK3 for phosphorylating substrates that have been prephosphorylated (or "primed") 4 amino acids C-terminal to the target Ser/Thr is due to the presence of a phosphate binding pocket in GSK3. The phosphate of the primed substrate sits in this pocket and positions the phosphate acceptor site to enable efficient phosphorylation by GSK3.
it was very intriguing when Klein and Melton discovered that the therapeutic agent lithium is a direct inhibitor of GSK3. This raised the hypothesis that inhibition of GSK3 is important in the therapeutic actions of lithium, as discussed in the following section, and provided the first selective inhibitor of GSK3 which greatly facilitated studies of the actions of GSK3. Because of the likely importance of lithium’s inhibitory effect on GSK3 in treating bipolar disorder, and because of GSK3’s potential involvement in other prevalent diseases, including Alzheimer’s disease and , during the last few years much effort has been focused on discovering new inhibitors of GSK3, several of which have been identified [12–14]. Thus, there are now several selective GSK3 inhibitors available and there is currently a large effort directed towards finding the potential therapeutic effects of GSK3 inhibitor.
Glycogen synthase kinase3 is emerging as a prominent drug target in the CNS
Glycogen synthase kinase3 (GSK3) is emerging as a prominent drug target in the CNS. The most exciting of the possibilities of GSK3 lies within the treatment of Alzheimer's disease (AD) where abnormal increases in GSK3 levels and activity have been associated with neuronal death, paired helical filament tau formation and neurite retraction as well as a decline in cognitive performance. Abnormal activity of GSK3 is also implicated in stroke. Lithium, a widely used drug for affective disorders, inhibits GSK3 at therapeutically relevant concentrations. Thus while the rationale remains testable, pharmaceutical companies are investing in finding a selective inhibitor of GSK3. In the present review, we summarize the properties of GSK3, and discuss the potential for such a therapy in AD, and other CNS disorders.
Abbreviations used
Aβ;β-amyloid
AD;Alzheimer's disease
CSF;cerebrospinal fluid
GSK3;glycogen synthase kinase3
MT;microtubule
NFT ;neurofibrillary tangle
NIDDM ;non-insulin-dependent diabetes mellitus
Glycogen synthase kinase 3 (GSK3) was initially discovered as a kinase involved in the regulation of glucose metabolism and later as a participant in wnt/wingless signaling. In mammals, two closely related isoforms GSK3α and GSK3β are present (Woodgett 1991). The GSK3β isoform is highly expressed in neural tissue where its expression is regulated during development.
GSK3 is also regulated by protein phosphatase 2A. GSK3 has multiple substrates including the tau protein implicated in AD and can phosphorylate both prephosphorylated and unmodified substrates. The main differences between GSK3α and GSK3β isoforms are found in the N- and C-terminal regions. Within the ATP pocket of GSK3 where most drugs bind to and compete with ATP, there appears to be only a single amino acid difference (Glu196 in GSK3α, Asp133 in GSK3β) making it difficult to identify an inhibitor that can distinguish the two isoforms.
GSK in AD
postmortem diagnosis of AD rests on the presence of extracellular plaques of β-amyloid (Aβ), and intracellular neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau protein. Increased levels of total GSK3 have not been consistently observed in AD brain, however, active GSK3 localizes to pretangle neurones, dystrophic neurites and NFTs in AD brain (Pei HYPERLINK "onlinelibrary.wiley.com/doi/10.1111/j.1471-4159.2004.02422.x/full"et alHYPERLINK "http://onlinelibrary.wiley.com/doi/10.1111/j.1471-4159.2004.02422.x/full". 1997). Neurons undergoing granulovascular degeneration also contain active GSK3β (Leroy HYPERLINK "onlinelibrary.wiley.com/doi/10.1111/j.1471-4159.2004.02422.x/full"et alHYPERLINK "onlinelibrary.wiley.com/doi/10.1111/j.1471-4159.2004.02422.x/full". 2002). A spatial and temporal pattern of increased active GSK3β expression coinciding with the progression of NFT and neurodegeneration has been observed. These studies provide evidence that the active form of GSK3β is increased in AD brain. Although a small increase in Aβ was observed when the β isoform of GSK3 was inhibited, non-isoform selective inhibitors such as lithium and kenpaullones as well as small interfering RNA against the α isoform of GSK3 reduce (Aβ) production in cells and in an animal model of amyloidosis (Phiel HYPERLINK "onlinelibrary.wiley.com/doi/10.1111/j.1471-4159.2004.02422.x/full"et alHYPERLINK "onlinelibrary.wiley.com/doi/10.1111/j.1471-4159.2004.02422.x/full". 2003). If this finding is consistent, GSK3 will gain significant importanceas a drug target for AD
Tau hyperphosphorylation in AD
;example Fig. 1).

Fig. 1 Model showing docking of Ser396–404 residues of Tau (arrow)
Tau microtubule associated protein is abundantly expressed in the brain and binds to tubulin through three or four repeat sequences in its C-terminal region to promote MT assembly. Abnormally phosphorylated tau forms filaments in vitro and in cells. In AD brains it accumulates in the neuronal perikarya and processes as paired helical filaments (PHFs), the chief constituent of NFTs. Hyperphosphorylation of tau is thought to result in the destabilization of MTs giving rise to a ‘pretangle’ stage and subsequently loss of dendritic MT and synapses, cytoskeletal degeneration, and eventually cell death .Although multiple protein kinases can phosphorylate tau, GSK3 and cdk5 were the key kinases purified from MT from AD brain .GSK3 phosphorylates the majority of sites on tau that are abnormally phosphorylated in AD brain.
GSK3, neuronal death and plasticity. decreases acetylcholine (ACh) levels by inhibiting pyruvate dehydrogenase (PDH) activity.GSK3 mediated inhibition of PDH decreases the conversion of pyruvate to acetyl Co-A, a precursor for the synthesis of Ach. GSK3 is upstream of the pro-apoptotic protease caspase-3. As active GSK3 triggers events that participate in cell death, part of AD pathology could result from an increase in activity of GSK inhibitor.GSK3 in neuroblastoma cells inhibits transcriptional. GSK3 also phosphorylates the transcription factor NFATc4 leading to a decrease in NFAT-regulated genes such as IP3R, which is required for neuronal plasticity. Thus negative regulation of CREB and NFATc4 by GSK3 could result in impaired synaptic plasticity, and cellular survival.
GSK3 and other CNS disorders treatment raising the possibility that GSK3 inhibition ers gene regulation and alters neuronal plasticity Chronic lithium treatment results in increased protein level of GSK3 substrate, b-catenin, in rat brain.
GSK3 inhibitors
inhibitors are paullones, indurubin anilinomaleimides 1.(thiadiazolidinones, and other small molecules. However, these inhibitors do not appear to be selective against the closely related kinases cdk2 and cdk5.
2.Indirubins inhibit GSK3 by competing with ATP but are not specific for GSK3 as they also inhibit cyclin dependent kinases (cdk).
2.Glaxo SmithKline’s maleimide derivatives,SB-415286 and SB-216763 also inhibit GSK3 activity by competing for the ATP binding site The thiadiazolidinones are ATP-non-competitive GSK inhibitors although these results will need confirmation. Most recently, a potent and specific inhibitor of GSK3 has been reported (AR-A014418) by AstraZeneca which is selective against cdk2, cdk5 and other kinases tested.
Pros and cons
The current therapeutics for AD provides marginal benefit by inhibiting the enzyme acetylcholinesterase and increasing the levels of the neurotransmitter ACh. Unfortunately, this type of therapy does not stop the progressive neuritic dystrophy and damage, and over time these therapies become ineffective.
Complete inhibition of GSK3 is not desirable; and could lead to potential side effects; a small inhibition to reset the stoichiometry of tauphosphorylation may be sufficient. It is also unclear whether a brief or sustained inhibition of GSK3 is necessary to attenuate tau phosphorylation for an extended period of time. 89,..[14,15,]s
AIMS AND OBJECTIVE
• To know all information about present Glycogen synthase kinase targeted drug and their action.
• To know about limitation of present Glycogen synthase kinase inhibitor.
• To find out more potent drug present till today with its action, side effect, limitation, and mode of action.
• To provide all information to those who are working on research in developing Glycogen Synthase kinase inhibitor.
DETAILS OF STUDY
Enzyme inhibitor is drug or compound which bind to the enzyme at its active site and inhibit its action by varios pathway Glycogen synthase kinase 3 regulates glycogen synthase, the rate-determining enzyme for glycogen synthesis.
GSK is enzyme which bind to the different receptor in CNS like NMDA ,Benzodizepam and GABA nergic Receptor,and its stimulation causes abnormal disorders in CNS like schizophrenia,bipolar disorders,mood disorders,Alziemer disorders,etc
So, we need to inhibit these enzyme in CNS by some inhibitor which use in treating CNS disorder.
As there is lot of recent literature dealing with the involvement of GSK-3 in the molecular pathways of different diseases, this review is mainly focused on the new GSK-3inhibitors discovered or speci?cally developed for this enzyme, their chemical structure, synthesis, and structure–activity relationships, with the aim to provide some clues for the future optimization of these promising drugs.
STORY
Glycogen synthase kinase has been implicated as a key player in several diseases: Alzheimer's, Huntington's, and possibly depression. Based on these relations, its potential market value as a possible drug target is obviously quite significant. From a patent perspective, GSK3β could offer novel drug classes for unique therapies in several disease areas where there are serious unmet medical needs;. It offers novel therapeutic strategies for CNS related diseases as well: Glycogen synthase kinase 3: a drug target in treatment of CNSHYPERLINK "http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15189333" HYPERLINK "http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15189333"disorder .
PATHWAYS
GSK works through several different pathway systems:
1)CNS Processes: In CNS tissues, GSK3 promotes amyloid Aβ formation by phosphorylating gamma-secretase complex, the last cleavage step for amyloid Aβ production. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides.In addition, GSK3 promotes Tau filament formation by phosphorylating the Tau subunits. PS1 activates PI3K thus inhibiting GSK-3 activity and tau over-phosphorylation: effects of FAD mutations.
COMPOUNDS
Several selective compounds have already been created and identified that function as GSK3 antagonists :
ARA014418 AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test.
Azakenpaullone 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3beta bis-7-indolylmaleimide Design, synthesis, and biological evaluation of novel 7-azaindolyl-heteroaryl-maleimides as potent and selective glycogen synthase kinase-3beta (GSK-3beta) inhibitors.
These inhibitors work on GSK3 by binding either competitively to the ATP-site (Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418) or non-competitively to the primer recognition site (a-4-Dibromoacetophenone, TDZD-8, OTDZT, 2-Chloro-1-(4,5-dibromo-thiophen-2-yl)-ethanone), thereby avoiding the typical non-selective problems associated with ATP-binding site antagonists In this regard, GSK3 is unique as a kinase target since new classes of drugs may be developed that avoid kinase ATP-site side effects, making them potentially quite selective. Some of the known inhibitors interact very little or not at all with CDK or PKA kinases. Interestingly, lithium also appears to inhibit via the priming site of GSK3, and may explain some of the CNS related benefits from lithium-based treatments. Lithium can therefore be used in a selective cellular assay to confirm GSK3 specificity.
GOALS
Find compounds that have excellent selective GSK3 IC50, with low binding to CDK's and other PK's, good permeability, and no tox metabolite.
Be sure to characterize mechanisms of action as well as possible for each of the disease areas with regard to GSK3 inhibition.
Compile different tox-response signatures on lead compounds by applying toxicogenomics.
Also assess the activities across different therapeutic spaces, keeping in mind different NOAEL (no-observed-adverse-effect-level) and age group (20-70, 40+, 60+) dependencies for different disease areas. This will have an impact on the allowable levels and variance of toxicity for later stages of drug development.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
STRATEGY
Since more knowledge is required about GSK3 role in different processes and tissues, we need to not only evaluate compounds based on binding affinity (IC50) to GSK, but to develop a profiling strategy to assess as many facets of GSK3 druggability for each of the therapeutic areas. Without this knowledge, many avenues of drug development may end in irremediable adverse effects and low efficacy.
A glycogen synthase kinase 3-beta promoter gene single nucleotide polymorphism is associated with age at onset and response to total sleep deprivation in bipolar depression.
Generate series of compounds based on literature-based inhibitors and newly constructed libraries. Focus on developing and enhancing high-throughput assays to measure non-competitive interactions with priming binding sites Identification of sequence variants and analysis of the role of the glycogen synthase kinase 3 beta gene and promoter in late onset Alzheimer's disease.
It is also known that GSK-3 is a key component of the Wnt signaling pathway, which is a central process at many stages of development and is highly conserved between Molecular cloning revealed that there were two closely-related isoforms, GSK-3aandGSK-3b.
Today, it is known that GSK-3 phosphorylates, and thereby, regulates the functions of manymetabolic, signaling, and structural proteins.GSK-3 has been linked to all the primary abnormalities associated withAlzheimer’s disease.
These include interactions between GSK-3 and components of the plaqueproducing amyloid system,the participation of GSK-3 in phosphorylating the microtubule-binding protein tau that may contribute to the formation of neuro?brillary tangles,and interactionsof GSK-3 with presenilinand other Alzheimer’s disease-associated proteins.
GSK-3 alsoregulates cell survival, as it facilitates a variety of apoptotic mechanisms.Thus, GSK-3 has acentral role in the regulation of the neuronal plasticity, gene expression, and cell survival, and may,be a key component of certain psychiatricand neurodegenerative diseases.Speci?city (such as glycogensynthase or tau protein) and mechanism of regulation of GSK-3. GSK-3 activity is negativelyregulated by phosphorylation on Ser 9 and positively regulated by phosphorylation on Tyr 216.
The amino acids involved in the primed substrate site of theenzymeor those implicated in the axin-binding sitethis kinase have been recently identi?ed.So nowadays, GSK-3 emerged in the medicinal chemistry research ?eld, as one of the most attractive therapeutic target for the development of selective inhibitors as new drugs, which may have hightherapeutic potential for the treatment of stroke, and neurological diseases such as bipolar disorders or Alzheimer’s disease.
As there is lot of recent literature dealing with the involvement of GSK-3 in the molecular pathways of different diseases, this goal is mainly focused on the new GSK-3 inhibitors discoveredor speci?cally developed for this enzyme, their chemical structure, and structure–activity relationships, with the aim to provide some clues for the future optimization of these promising drugs.
MOOD DISORDERS AND GSK
Bipolar affective disorder, in which patients have a history of experiencing manic episodes that are often interspersed with depression, and major depression are commonly referred to as mood disorders.. Research into the causative mechanisms has been greatly hampered by the lack of adequate animal models of these diseases. However, investigations of the mechanisms of action of therapeutic agents have provided leads about possible pathological mechanisms, and recent findings have revealed a number of connections linking GSK3 to the causes and, especially, to the actions of therapeutic agents used in these disorders.
For approximately the last fifty years the drug lithium has been the mainstay for the treatment of bipolar disorder, with a beneficial effect often observed in approximately 60–80% of patients and with no tolerance or sensitivity developing throughout many years of treatment. GSK3 was first linked to bipolar disorder in 1996 by the finding that lithium is a direct inhibitor of GSK3. They found that lithium inhibited GSK3 with an IC50 of approximately 2 mM, slightly greater than the therapeutic concentration range of lithium in serum, which is approximately 0.5–1.5 [1]. Soon thereafter, GSK3 was shown to be inhibited by lithium both in intact cells and in mammalian brain in vivo. These and subsequent examinations of many kinases confirmed that the inhibitory effect of lithium was a relatively selective action targeted to GSK3.
The discovery that lithium directly inhibits GSK3 introduced the concept that this action may contribute to the therapeutic effects of lithium in mood disorders and, consequently, that GSK3 may be dysregulated in mood disorders. However, because the therapeutically effective concentration of lithium in serum is lower than the IC50 of lithium for inhibition of GSK3, it seemed that the inhibition of GSK3 by lithium at therapeutic levels may be too little to contribute significantly to mood stabilization. A solution to this limitation was provided by the discovery of an in vivo amplification mechanism for lithium’s inhibition of GSK3 .
We found that chronic (4 weeks) in vivo treatment with a therapeutically relevant regimen of lithium administration increased by several-fold the phosphorylation of serine-9 of GSK3β in mouse brain regions.Increased serine-phosphorylation of GSK3 following lithium administration indicates that the modest direct inhibitory effect of lithium on GSK3 is amplified by this phosphorylation mechanism, providing more substantial inhibition of GSK3 at a therapeutically relevant concentration of lithium than would be attained only by the direct inhibitory effect of lithium. These observations raised the exciting prospect that lithium inhibits GSK3 with amplification-mediated selectivity in the magnitude of inhibition .
Other therapies used for mood disorders also have been linked to inhibition of the activity of GSK3.
Valproic acid, originally used as an anticonvulsant and now also widely used as a mood stabilizer in bipolar disorder, was reported to directly inhibit GSK3 activity by some investigators but not others.Valproic acid treatment also increased the inhibitory serine phosphorylation of GSK3 in human neuroblastoma cells.
Although in vivo treatment with valproate did not increase serine phosphorylation of GSK3, valproate treatment did reduce pathophysiologically-induced dephosphorylation of both isoforms of GSK3. Thus, like lithium, valproate appears to contribute to the inhibitory control of GSK3 in mammalian brain in vivo. Increased serine phosphorylation of GSK3β in mouse brain also was induced by electroconvulsive seizure treatment of mice, another effective and widely used therapeutic intervention for bipolar disorder. Thus, it is intriguing to find that three disparate mood stabilizing therapies, lithium, valproic acid, and electroconvulsive seizures, have the common action of causing inhibition of GSK3. These findings support the postulate that inhibition of GSK3 contributes to the therapeutic actions of mood stabilizers.
In summary, evidence that the actions of GSK3 are involved in the pathophysiology of bipolar mood disorder stems from the inhibitory actions on GSK3 of therapeutic interventions used in this illness. This connection should not be over-interpreted to indicate that GSK3 itself is hyperactive in bipolar disorder. Although this is one possibility, it is equally likely that upstream or downstream signals linked to GSK3 may be altered in bipolar disorder. For example, signaling activities upstream of GSK3 may be the primary deficiency, resulting in inadequate inhibitory control of GSK3 by these signaling pathways which would be bolstered by these therapies that inhibit GSK3.
Alternatively, targets downstream of GSK3 may be dysfunctional, so these may be bolstered by inhibition of GSK3 by these therapies. Thus, either GSK3 itself or signaling intermediates coupled to GSK3 may be abnormal in bipolar disorder, so inhibition of GSK3 may contribute to normalizing these targets.
Dysfunctional GSK3, or molecules linked to GSK3, also may contribute to major depression. The most prevalent model for the pathological cause of depression is the monoamine hypothesis, which suggests that depression stems from inadequate serotonin (5HT) and/or norepinephrine neurotransmission. This hypothesis is supported by the findings that most antidepressants facilitate mono-aminergic neurotransmission, especially serotonergic actions, although the disease is clearly more complex .Therefore, much research is focused on identifying intracellular signaling pathways coupled to 5HT receptors that may be involved in mood disorders and that may provide new targets for therapeutic agents. If key signaling outcomes downstream of receptors can be identified, then drugs targeting these signaling pathways may provide a therapeutic approach that is an alternative, or an add-on, to classical antidepressants.
Recent evidence indicates that lithium’s inhibition of GSK3 may fulfill such a role, since GSK3 was recently found to be a downstream target in 5HT receptor-mediated signaling pathways that may not be adequately inhibited in depression (discussed below), and the GSK3 inhibitor lithium can be an effective add-on agent in antidepressant-refractory depression.
The deficiency in serotonergic activity in depression makes especially relevant recent findings that serotonergic activity contributes to the inhibitory control of GSK3 in mammalian brain in vivo, so that serotonergic deficiency would lead to abnormally activated GSK3. In this study, serotonergic activity was increased in vivo by administration of d-fenfluramine and clorgyline to mice.
D-fenfluramine induces the release of 5HT and inhibits 5HT reup-take, and clorgyline is a monoamine oxidase inhibitor that inhibits the breakdown of 5HT. Therefore, administration of d-fenfluramine and clorgyline augments 5HT levels, and this was found to increase the inhibitory serine-9 phosphorylation of GSK3β in mouse pre-frontal cortex, hippocampus, and striatum .Increasing 5HT levels by inhibition of monoamine reuptake using the antidepressants fluoxetine or imipramine also increased serine-9 phosphorylation of GSK3β in mouse brain. These results demonstrate that increased serotonergic activity following the administration of anti-depressants inhibits GSK3β in brain.
Examination of 5HT receptor subtypes showed that stimulation of 5HT1A receptors in vivo caused increases in serine-9 phosphorylation of GSK3β .Conversely, blockade of 5HT2 receptors by administration of a selective antagonist increased the serine-9 phosphorylation of GSK3β , indicating that 5HT2 receptors normally cause dephosphorylation of phospho-Ser9-GSK3β (Fig. 3). This balance of 5HT1A and 5HT2 receptors in regulating the phosphorylation of GSK3 is an interesting finding, since much previous evidence suggests that an imbalance between 5HT1A and 5HT2 receptors is associated with depression.
studies have provided evidence that 5HT1A receptors are deficient in major depressive disorder and that 5HT2 receptors are up-regulated in depression, although this remains a subject of intense investigation Thus, these alterations would lead to decreases in the inhibitory serine phosphorylation of GSK3 in depression. It has been hypothesized that antidepressants exert their therapeutic effects through restoring the balance between those two receptor subtypes ,which may restore phospho-Ser9-GSK3β to normal levels.
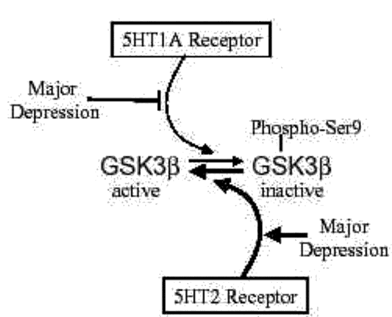
Fig 3:Schematic depiction of the regulation of GSK3 by 5HT1A and 5HT2 receptors and changes that may be associated with major (20)
GSK3β is inhibited by phosphorylation of serine-9. This inhibitory phosphorylation is promoted by activation of 5HT1A receptors. However, decreased 5HT1A receptors occur in major depression, so there may be insufficient signaling leading to inhibition of GSK3β in depression. Conversely, activation of 5HT2 receptors cause activation of GSK3β by promoting its dephosphorylation. Increased 5HT2 receptors occur in major depression, suggesting increased activation of GSK3β . Overall, the balance between 5HT1A and 5HT2 receptors contributes to maintaining the normal activity level of GSK3β , and changes in each receptor associated with depression disrupt the 5HT receptor-mediated inhibitory signals that normally control GSK3β , suggesting that the activity of GSK3β is not adequately controlled in major depression.
The signaling pathways linking serotonergic activity to the regulation of phospho-Ser-GSK3 in brain in vivo are not known since many pathways converge on GSK3 and it is difficult to make mechanistic conclusions based on in vivo studies where many signaling pathways converge on GSK3. Most serotonergic receptors are coupled to classical second messenger pathways by one of three types of heterotrimeric G-proteins, Gq which couples receptors to the phosphoinositide second messenger system, and Gi or Gs which couple receptors to inhibition or activation, respectively, of cyclic AMP production. Gq-coupled serotonergic receptors activate protein kinase C which, in turn, is known to phosphorylate the regulatory serine of GSK3 .We are unaware of any reports that directly demonstrate protein kinase C-mediated serine phosphorylation of GSK3 following serotonergic receptor activation although this connection seems likely to occur. Gi-coupled receptors are known to activate the PI3K/Akt signaling pathway, so Akt activated in this manner could account for serine phosphorylation of GSK3 by increased serotonergic activity. 5HT1A receptors coupled to Gi have been shown to activate PI3K and Akt .Furthermore, the activity of Akt in brain samples from depressed suicide victims were below that of matched controls .This indicated that depression might be associated with diminished activity of the PI3K/Akt signaling pathway which normally leads to inhibition of GSK3β , supporting the possibility that GSK3β may not be adequately inhibited in depression.
The hypothesis that hyperactive GSK3 may partially contribute to depression or behaviors associated with depression was further supported by studies of the effects of administration of GSK3 inhibitors to mice. The most intriguing aspect of these studies are results using the forced swim test, a widely used model to assess depressive behavior and the effects of antidepressant agents, measured by immobilization time which is diminished by most antidepressants. Kaidanovich-Beilin et al. [46] found that administration of a peptide inhibitor of GSK3β rapidly induced antidepressant-like behavioral effects, specifically reducing immobilization in the forced swim test. That this depression-associated behavior is modulated by GSK3 was further supported by recent studies [47, 48]. Gould et al. [48] reported that administration of an ATP competitive GSK3 inhibitor modestly reduced immobilization in the forced swim test. O’Brien et al. showed more directly the relation between GSK3β and immobilization in the forced swim test. They found that large reductions in immobilization time were induced not only by inhibition of GSK3β by lithium administration, but also that lowered GSK3β levels expressed in heterozygote GSK3β +/− mice was associated with a large reduction of immobilization in the forced swim test. Taken together, these three reports demonstrate convincingly that GSK3 has a critical role in this widely used paradigm to assess depressive activity and the counteracting effects of antidepressants.
SCHEZOPHRENIA AND GSK
Several lines of research have produced findings consistent with the hypothesis that alterations in GSK3 are connected with schizophrenia. However this association has not received the same intense scrutiny as it has in mood disorders, and contradictory findings have been reported, so the association of GSK3 with schizophrenia, while tantalizing, remains to be more thoroughly evaluated.
Schizophrenia is a prevalent and severe psychotic disorder with considerable variation among individuals in symptoms associated with thought content, perception, cognition, and affect. The causative factors of schizophrenia remain unknown, but dysregulated dopamine neurotransmission is likely the most widely investigated hypothesis of the pathophysiology of schizophrenia. This classical hyperdopaminergic hypothesis of schizophrenia pathology is supported by the therapeutic effects in schizophrenia of conventional antipsychotics that are dopamine D2 receptor antagonists and by the psychotogenic effects of dopamine enhancing drugs. However, although conventional antipsychotics can diminish symptoms in some patients, it is evident that the pathophysiology of schizophrenia is more complex, and diverse, than being caused only by increased dopaminergic activity in subcortical regions.
Some symptoms of schizophrenia, such as cognitive impairments, are resistant to conventional antipsychotics, and the cognitive deficits in schizophrenia are thought to arise in part from hypodopaminergic neurotransmission at dopamine D1 receptors in the pre-frontal cortex . This is supported by clinical studies that show atypical antipsychotics, which increase dopamine neurotransmission at dopamine D1 receptors, improve cognitive symptoms in schizophrenic patients .Especially intriguing is the evidence showing that dopamine D1 receptor hypoactivity in the pre-frontal cortex can result in dopamine D2 receptor hyperactivity in the striatum. Imaging studies of the function of dopamine D1 receptors in the prefrontal cortex [56–58] and of dopamine D2 receptors in the striatum of schizophrenic patients lend further support to the view that an imbalance of cortical/subcortical dopaminergic function may be central to the pathology of schizophrenia.
Thus, regarding dopaminergic neurotransmission, balanced activities of dopamine D1 and D2 receptors seems to be critical, and schizophrenia appears to be associated with low dopamine D1 and/or high dopamine D2 receptor function. Beyond the dopaminergic system, many other theories of the pathology of schizophrenia have been promulgated.
One of the most widely considered is the evidence of neurodevelopmental abnormalities, supporting the concept that schizophrenia represents a spectrum of diseases with multi-factorial pathologies [49]. Although studies connecting GSK3 to schizophrenia are few, they have identified links between GSK3 and these two major postulates, altered dopaminergic activity and disrupted neurodevelopment.
Abnormalities of GSK3 were first linked to schizophrenia in a series of studies reported by Agam and colleagues. They found approximately 40% lower GSK3β mRNA levels, GSK3β protein levels, and GSK3 kinase activity in postmortem samples of frontal cortex from subjects with schizophrenia, and a 30% lower GSK3β protein level in the cerebrospinal fluid, compared with controls. However, these differences were not detected, except for the lower GSK3 mRNA level, by the same investigators in samples from a different brain collection. This difference between brain collections was also encountered by another group who found differences of GSK3β protein levels in schizophrenic compared to control samples in one brain collection but not another .
As noted earlier in this review, a lack of changes in GSK3 levels does not preclude changes in GSK3 actions because of the intracellular mechanisms that regulate its activity, such as the inhibitory effect of serine-phosphorylation, but this can not be studied in human brain samples because of the extensive loss of serine phosphorylation of GSK3 that occurs postmortem. Further studies with a greater number of samples will be necessary to draw concrete conclusions about whether or not alterations in GSK3 expression or protein levels reproducibly occur in subjects with schizophrenia.
Another approach to identifying potential links between schizophrenia and dysregulated GSK3 is to examine the modulatory influences of neurotransmitter systems that are thought to be involved in the illness, as discussed in the previous section concerning studies of the serotonergic system in mood disorders. Therefore, since there is evidence for dopaminergic dysregulation in schizophrenia, it is pertinent to consider whether dopaminergic activity has a role in regulating GSK3.
This was first examined in brain in vivo by Gil et al. who found that administration of a dopamine D1 receptor agonist inhibited GSK3 activity in rabbit frontal cortex and hippocampus, and this was blunted in rabbits following prenatal cocaine exposure which itself caused inhibition of GSK3β activity.
Thus, this study showed for the first time that dopaminergic activity has a regulatory influence on GSK3 in brain in vivo, and that long term changes in the dopaminergic system can modulate GSK3. From this initial report, especially notable is the possibility that low dopamine D1 receptor activation that is reported to occur in schizophrenia would be associated with impaired inhibitory control of GSK3 (Fig. 4).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
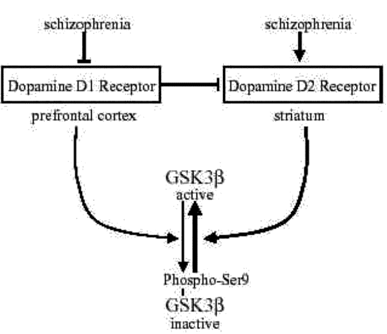
Fig 4:Schematic depiction of the regulation of GSK3 by dopamine D1 and dopamine D2 receptors and changes associated with schizophrenia
GSK3β is inhibited by phosphorylation of serine-9. This inhibitory phosphorylation is promoted by activation of dopamine D1 receptors. However, decreased dopamine D1 receptor activation occurs in schizophrenia, so there may be insufficient signaling leading to inhibition of GSK3β in schizophrenia. Conversely, activation of dopamine D2 receptors cause activation of GSK3β by promoting its dephosphorylation. Increased dopamine D2 receptor activation occurs in schizophrenia, in part by deficient inhibitory inputs from dopamine D1 receptors, suggesting increased activation of GSK3β . Overall, the balance between dopamine D1 and D2 receptors contributes to maintaining the normal activity level of GSK3β , and changes in each receptor associated with schizophrenia disrupt the control of GSK3β .
Subsequently, more extensive in vivo interactions between the dopaminergic system and GSK3 in mammalian brain were reported. In a recent definitive study, Beaulieu et al. found that increased dopaminergic stimulation of dopamine D2 receptors in the striatum induced by administration of the indirect dopamine stimulant amphetamine or present in dopamine transporter knockout mice (DAT-KO), caused activation of GSK3α and GSK3β in mouse striatum. This appeared to occur because of decreased Akt activity which resulted in decreased serine-phosphorylation of GSK3. Hyperactive GSK3 was shown to contribute to the behavioral phenotype because administration of GSK3 inhibitors, including lithium, antagonized dopamine-dependent hyperactivity and stereotypy in the DAT-KO mice, and amphetamine-induced hyperactivity was lower in GSK3β +/− mice.
These findings clearly demonstrated that GSK3 is a downstream target of dopamine D2 receptor-mediated signaling in vivo and that GSK3 mediates some of the behavioral effects of dopamine, supporting the possibility that alterations in GSK3 activity may be relevant to schizophrenia and other dopamine-related disorders .This study notably raises the possibility that GSK3 is abnormally activated in schizophrenia since dopamine D2 receptor activation is elevated, which in conjunction with impaired dopamine D1 receptor-mediated inhibition of GSK3 may synergistically contribute to hyperactivated GSK3 (Fig. 4).
More direct evidence of impaired Akt/GSK3β signaling in subjects with schizophrenia was recently reported. Emamian et al. [68] found approximately 50% decreases in the protein levels of one isoform of Akt, called Akt1, in the frontal cortex and lymphocytes of subjects with schizophrenia compared with controls. Administration to mice of haloperidol, a typical antipsychotic which is an antagonist of dopamine D2 receptors, increased the activating phosphorylation of Akt and the inhibitory serine-phosphorylation of GSK3β in brain.
The decreased Akt signaling to GSK3β in schizophrenia and corrective modulation by the dopaminergic antagonist supports the potential role of dopamine receptor-coupled signaling to Akt and GSK3β in the pathogenesis of schizophrenia. This study also reported lower phospho-Ser9-GSK3β levels in samples from subjects with schizophrenia compared with controls as determined by immunoblot analysis . However, the recently reported rapid postmortem serine-dephosphorylation of GSK3β suggests that more thorough examination of immunoreactive bands is necessary to unequivocally identify levels of phosphoserine-GSK3 in postmortem samples.
Most interestingly, Emamian et al. found that a haplotype of Akt1 that was preferably transmitted to schizophrenic probands is related to a lower protein level of Akt1 and that amphetamine administration to Akt1-depleted mice showed disruption of prepulse inhibition, a representative model of impaired sensorimotor gating of schizophrenia.
A recent report confirmed that Akt1 is a susceptibility gene for schizophrenia in a large population study but it was not confirmed in another study . This finding suggests that reduced Akt1 may contribute to schizophrenia, supporting the possibility of an association between impaired control of GSK3 and schizophrenia.
The role of GSK3β in association with the Wnt signaling pathway (Fig. 2A) is a well known factor regulating CNS development, so altered GSK3β signaling in the brain of subjects with schizophrenia also could contribute to the neurodevelopmental abnormalities that have been linked to schizophrenia. There have been several reports of alterations in the Wnt signaling pathway, which regulates the action of GSK3, associated with schizophrenia . Since Wnt signaling is a key component of neurodevelopment, and much evidence indicates neurodevelopmental abnormalities in schizophrenia , these are tantalizing reports, but the significance of these findings for the pathophysiology of schizophrenia remain to be investigated in greater detail.
In addition to dopaminergic activity, alterations of cholinergic and glutamatergic neurotransmission also have been linked to the pathology of schizophrenia, so it is of interest that each of these neurotransmitter systems recently was found to influence the regulation of brain GSK3 in vivo. Schizophrenia has been linked to dysregulated cholinergic neurotransmission in several studies, and especially strong is the evidence indicating association with the cognitive impairment of schizophrenia .
Cognitive impairment is often evident in schizophrenia, and schizophrenia has been reported to be associated with reduced choline acetyltransferase, the enzyme that synthesizes acetylcholine which is a critical neuro-transmitter for cognition, and choline acetyltransferase activity was reported to be inversely correlated with cognitive impairments in schizophrenia.
There have been several reports of decreased levels of muscarinic receptors in specific brain regions of schizophrenic patients, including frontal cortex anterior cingulate gyrus , hippocampus , Broadmann’s area , and caudate-putamen .
Taken together, these and other findings suggest that muscarinic receptor stimulation can be impaired in schizophrenia either at the level of acetylcholine synthesis or receptor activation. Therefore, it is of interest that a regulatory influence of cholinergic activity modulating the phosphorylation of brain GSK3 in vivo was recently identified. De Sarno et al. found that cholinergic stimulation with the muscarinic receptor-selective agonist pilocarpine or the acetylcholinesterase inhibitor physostigmine rapidly increased the serine-phosphorylation of GSK3α and of GSK3β by several-fold in three mouse brain regions. This finding raised the possibility that dysfunctional cholinergic activity may cause inadequate inhibitory control of GSK3 which can be restored by stimulation of muscarinic receptors.Much research has linked altered glutamatergic neurotransmission to schizophrenia .
One of the most widely used models of schizophrenia involves application of glutamatergic N-methyl D-aspartate (NMDA) receptor antagonists to animals because in healthy human subjects these agents can induce symptoms similar to those seen in schizophrenia . Thus, administration of the noncompetitive NMDA receptor antagonists phencyclidine or ketamine can induce several symptoms of schizophrenia in normal control individuals, and can worsen symptoms in schizophrenic subjects Conversely, administration of NMDA receptor agonists examined as adjunctive treatments have been reported to improve psychotic symptoms in schizophrenia . These findings support the hypothesis that activation of NMDA receptors may be impaired in schizophrenia.
This connection between NMDA receptor activity and schizophrenia raises the question of whether this may contribute to the regulation of GSK3, and several recent studies have provided support for this regulatory interaction. NMDA treatment of cultured hippocampal neurons caused a rapid and nearly complete dephosphorylation of phospho-Ser9-GSK3β , indicating that GSK3β is activated by NMDA receptor signaling. In accordance with that conclusion, in vivo blockade of NMDA receptors by administration of the antagonist phencyclidine increased mouse brain serine-phosphorylation of GSK3, a response was also observed in mouse brain following administration of memantine, an NMDA antagonist approved for use in humans.A conflicting report showed that in immature rats blockade of NMDA receptors by in vivo administration of the antagonist MK-801 transiently decreased the serine-phosphorylation of GSK3 , a difference from the other reports that could be due to age-dependent differences in responses to NMDA receptor modulation or differences between NMDA antagonists. Thus, although still only few, the majority of studies indicate that NMDA receptor stimulation dephosphorylates GSK3 and that blockade of NMDA receptors in vivo is sufficient to cause increased levels of serine-phosphorylated GSK3.
Overall, quite a few connections have been identified between GSK3 and schizophrenia, but there are also serious contradictions in this data. Thus, some data suggests the action of GSK3 is reduced, whereas other data suggests it is increased, in association with schizophrenia. Schizophrenia-associated reductions of GSK3 are indicated by the measurements in postmortem brain samples and by the inhibitory effects of the NMDA antagonists phencyclidine and memantine. Postmortem measurements are the most direct approach to identifying disease-related links, but this strategy is also fraught with difficulties inherent in using postmortem tissue and in studying such a heterogeneous sample population. These difficulties are exemplified by the mixed results obtained in different sample sets. Countering the indications of reduced GSK3, there is substantial evidence of increased GSK3 actions in schizophrenia. This evidence comes from studies showing reduced Akt in schizophrenia and studies of neurotransmitter effects on regulating GSK3. The two studies implicating Akt deficits in schizophrenia provide strong evidence that this inhibitory regulator of GSK3 is dysfunctional, thus allowing hyperactivation of GSK3. This is corroborated by some indications in schizophrenia of low dopamine D1 receptors which inhibit GSK3, and elevated dopamine D2 receptors that activate GSK3, and that typical antipsychotics block D2 receptors, which would cause inhibition of GSK3. The altered balance of D1 and D2 receptors, along with a possible deficit in cholinergic neurotransmission, lead to the prediction that GSK3 is inadequately controlled in schizophrenia. However, lithium, a GSK3 inhibitor, has very limited therapeutic effects in schizophrenia, suggesting that if GSK3 activity is abnormal in schizophrenia it may only contribute to a subset of symptoms. Thus, although intriguing connections between schizophrenia and alterations of GSK3 have been identified, much more research is necessary to integrate the findings from studies of GSK3 in postmortem tissue, the developmental influences of GSK3, and the regulatory effects on GSK3 of neurotransmitter systems that have been shown to be involved in U
FUNCTION REGULATED BY GSK WHICH MAY UNDERLINE THE PATHOLOGY AND TREATMENT MECHANISM IN PSYCHIATRIC DISEASES
The evidence linking GSK3 to the pathology and treatment of mood disorders and schizophrenia raises the key question of how dysregulated GSK3 might contribute to these diseases. As the understanding of the actions of GSK3 has been greatly expanded during the last few years, several effects of GSK3 have been identified as strong candidates that might link its dysregulation to these diseases. These actions are centered on neural plasticity, and this is considered below within the contexts of structural effects, neurogenesis, gene expression, and responses to stress.
Plasticity: Cell Structure and Remodelling
Perhaps the most widely accepted conceptual basis for mood disorders, and also considered in schizophrenia research, is the postulate that there is impaired neural plasticity associated with the pathophysiology of these diseases. One reason for the fairly widespread acceptance of this concept is that it can be applied to nearly every aspect of neuronal function, thus its application to mood disorders really does little to help to precisely and specifically define key functions that are dysregulated in these diseases. Therefore, for the purpose of this review, we define neural plasticity more narrowly as the ability of neurons to respond appropriately to fluctuating inputs, such as changes in neurotransmission, stress, or cellular insults. In other words, these represent adaptive changes that facilitate neuronal function in response to alterations in external inputs, and these inputs may be normal fluctuations in neurotransmission or exposure to stress. In this context, much evidence has documented the crucial role of GSK3 in neuronal plasticity, and here we consider specifically structural plasticity as an example of one of the many influences of GSK3 on neural plasticity.
GSK3 has many influences on cell biology and architecture, as recently reviewed [5]. Briefly, GSK3 phosphorylates several proteins that can bind to microtubules, protein complexes that provide structural stability to neurons but which must maintain plasticity in order to allow the dynamic changes in neuronal shape and contacts which continually occurs in neurons. These substrates of GSK3 include the microtubule-associated protein (MAP) tau, MAP1β , APC, CRMP-2, and others. By phosphorylating microtubule-associated proteins GSK3 modulates microtubule dynamics which are important in neuronal remodelling, neurite outgrowth and collapse, and axonogenesis [97–104]. These are all key processes in structural neural plasticity, emphasizing the widespread influences of GSK3 and the importance of maintaining strict control of GSK3 activity. GSK3 also phosphorylates the protein motor kinesin, thereby modulating intracellular transport of many types of cargo [105], modulates growth cone extension [106, 107], and modulates cell motility [108]. Thus, fluctuations in GSK3 activity modulate many intracellular structural dynamics of neurons. GSK3 is also an essential component of several developmentally important signaling pathways (which are also functional in adult brain), including Wnt [6, 7], Hedgehog [109, 110], Reelin [111], and Notch signaling, each of which controls aspects of neuronal structure. Dysregulation of GSK3 associated with these developmental systems may be particularly relevant to schizophrenia for which there is a strong body of evidence of developmental deficiency. Thus, GSK3 has numerous effects on cell biology, architecture, and remodeling, actions that may underlie its detrimental effects in psychiatric disorders when it is not properly regulated. For example, deficient serotonergic activity, as can occur in mood disorders, can cause GSK3 in the brain to be abnormally active. This can impair neural plasticity through the actions of GSK3 on these dynamic structural targets, and bolstering serotonergic activity with antidepressants is now known to strengthen the inhibitory control of GSK3 in the brain, thereby potentially facilitating neural plasticity by modulating these structural dynamics.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Neurogenesis
Impaired neurogenesis in animal models of depression, and its correction by antidepressants, has recently brought this process into prominence as a potential crucial mechanism in depression and other psychiatric diseases. Neurogenesis in this context involves the production, survival, or integration of new neurons in the adult brain. In a simplified manner, this theory posits that depression may be causally associated with impaired neurogenesis, and that antidepressants augment one or more of the components of neurogenesis. Thus it is intriguing that GSK3 also is a noted regulator of neurogenesis. Using a variety of experimental systems, several early studies nearly simultaneously reported increases in markers of neurogenesis following diverse treatments in animals or cells with agents that are therapeutic in mood disorders, including antidepressants, electroconvulsive shock, and lithium. None of these studies with lithium examined the mechanism by which neurogenesis was enhanced, but studies of GSK3 make it a target worth investigating. For example, in addition to the well known role of the Wnt signaling pathway (activation of which inhibits GSK3) on neuronal development ,several studies reported effects of inhibitors of GSK3 on neurogenesis in embryonic stem cells,.Thus, inhibition of GSK3, which may be impaired in mood disorders, by lithium and other therapeutics may bolster neurogenesis, an action that may contribute to therapeutic outcomes. With respect to depression, especially compelling is the recent report by Santarelli et al.]which provided strong evidence that the behavioral effects of chronic antidepressants may be mediated by the stimulation of hippocampal neurogenesis? Since serotonergic activity is reduced in depression, it is also of interest that several studies found activation of 5HT1A receptors, which we showed [36] causes phosphorylation (inactivation) of GSK3, enhanced neurogenesis. In summary, there is growing, but still very incomplete, data suggesting links between neurogenesis and both mood disorders and therapeutic agents, and that GSK3 may contribute to these regulatory mechanisms
(16,17,18,19)
CLASSIFICATION OF GLYCOGEN SYNTHASE KINASE INHIBITORS ACTING IN CNS,
1.Peptide inhibitor.
2.Mood stabilizer drug.
3.Small molecule targeting glycogen synthase kinase
• Azole and Related compound.
• Micellaneous heterocyclic compound.
1.Purin and pyrimidine derivatives
2.Indole derivatives
• . Maleimides derivatives
• MuscarinicAgonist
• f . ATP noncompetitive inhibitor
1.Peptide inhibitor
Dipeptidyl peptidase-4 inhibitors(DPP-4 inhibitors) are enzyme inhibitorsthat inhibit the enzymedipeptidyl peptidase-4(DPP-4) and are a potenttreatment for type 2 diabetes
2.Mood Stabilizer
LITHIUM, THE FIRST GSK-3 INHIBITOR
Lithium is the primary therapeutic agent for bipolar mood disorder., previously, lithium has been discovered as a selective inhibitorof GSK-3,33–36which has contributed extensively to the study of the signaling pathways of thisenzyme. On the other hand, this discovery raises the possibility that disregulation of GSK and itsinhibition by lithium may contribute to the mental disorder and its treatment.
MODE OF ACTION
the mechanism by which lithium (Liþ) inhibit GSK is recent hypothesis are recently proposed.
1. Competitive inhibition of GSK by lithium with respect to magnesium by potassium deprivation,
2. other one proposes (Mg2þ), but not to substrate or ATP.
Given the cellular centrations of ATP and Mg2þ, these results indicate that Li þwill have a greater effect on GSK-activity in vivothan expected from in vitrostudies, and this may be a factor relevant to its use in the treatment of depression.Among the multiple effects in the brain that lithium has, the inhibition of GSK-3 induced tau phosphorylation is demonstrated. Lithium reversibly reduced tau phosphorylation at therapeutic concentrations, and even at high concentrations did not alter the neuronal morphology. Additionally, lithium prevents the neuronal death induced by the ?brillaryb-amyloid and neuro-protection appers to be mainly due to GSK-3 inhibition.These results carry implications for futurestudies of the actions of mood-stabilizing drugs and indeed of the molecular mechanisms of affective disorders.In this sense, another mood-stabilizing drug,
valproate,
therapeutic use also used in treating bipolar disordersand epilepsy, has been shown to inhibit GSK-3 enzyme.
MODE OF ACTION
Valproic acid
Valproic acid, however, acts through a distinct pathway that involves direct inhibition of histone deacetylase.
Side effectmechanism for valproic acid-induced birth defects, and could also explain the ef?cacy of valproic acid in the treatment of bipolar disorders.
3.SMALL MOLECULES AS GSK-3 INHIBITORS
Regarding the high therapeutical potential of targeting GSK-3 in many different pathologies, the search for its inhibitors is a very active ?eld in both academic centers and pharmaceuticalcompanies.
purines derivativesdeveloped by Chiron had been the ?rst synthetic molecules speci?cally reported as GSK-3 inhibitors.These heterocycles, which may be chemically prepared according to the method of Norman,47have inhibitory GSK-3 activity in vitro. The most active compound tested showed a 63% inhibition of GSK-3 activity at 1mM.But what is more frequent until now for discovering new GSK-3 inhibitors, are the screeningprograms speci?cally aim at ?nding GSK-3 inhibitory activity in other compounds previouslyreported with other biological properties. This is the case of the kinases inhibitors hymenialdisine, paullones, indirubines, and maleidimides or the m1muscarinic agonist initially developed ascholinergic drugs. Finally, a special mention to the thiadiazolidinones (TDZD) molecules as the ?rst non-ATP competitive GSK-3 inhibitors.
a.Azole and Related compound(20)
Hymenialdisine
MODE OF ACTION:
The marine sponge constituent hymenialdisine is a potent inhibitor of cyclin-dependent kinases (CDK’s), casein kinase 1 and GSK-3. Hymenialdisine competes with ATP for binding to all these kinases. The CDK2-hymenialdesine complex crystal structure shows that three hydrogen bonds link hymenialdesine to the Glu-81 and Leu-83 residues of CDK2. This compound inhibits GSK-3 as shown by the inhibition of MAP-1B phosphorylation.Hymenialdisine also blocks the in vivophosphorylation of the microtubule-binding protein tau at sites that are hyperphosphorylated by GSK-3band CDK5/p35 in Alzheimer’s disease. For this reason, the natural product, hymenialdesine, is a new kinase inhibitor with promising potentialapplications for treating neurodegenerative disorders. Additionally, it is known that GSK-3 is required for nuclear functioning of NF-kB49and impliesthat small molecules inhibitors of GSK-3 should be potent anti-in?ammatory agent.
d.Micellaneous heterocyclic compound.
1. Purins and pyrimidine derivatives
Paullones
Paullones constitute a new family of benzazepinones with promising anti-tumoral properties.They have been recently described as potent, ATP competitive, inhibitors of the cell cycle regulating CDK’s.51Kenpaullone was identi?ed as a potent inhibitor of CDK’s when ?avopiridol) Kenpaullone was docked to the ATP binding site of the CDK2 structure de?ning the key structural features for effcient interaction. It was postulated that substituting a hydrogen bond acceptor at 9-position could increase af?nity by forming hydrogen bond with a water molecule observed in thecrystal structure of CDK2 and ATP bound. Moreover, this type of substituent could enhance af?nity by increasing the strength of the hydrogen bonds to Leu-83 and increasing the strength of the interactions with the hydrophobic side chains by rendering the ring systems relatively charge de?cient. Structure–activity data con?rmed this hypothesis being the 9-nitro paullone (alsterpaul-lone5), the most active compound synthetized; exceeded the CDK1 inhibitory potency of?avopiridol (IC50CDK1/Cyclin B¼0.035mM) and exhibited a remarkablein vitroanti-tumoractivity.
Alsterpaullone
paullones have been proved to be potent CDK5/p25 inhibitors Alsterpaullone was demonstrated to act by competing with ATP for binding to GSK-3, inhibits the phosphorylation of tauin vivoat sites, which are typically phosphorylated by GSK-3 in Alzheimer’s disease and also inhibits the CDK5/ p25-dependent phosphorylation of DARPP-32 in mouse striatum slicesin vitro
2.INDOLE derevatives
30monoxime, which is over 10-fold less active on GSK-3bthan CDKs.
As CDK5 and GSK-3bare two major kinases involved in tau hyperphosphorylation,59indirubins and many reported CDK’s inhibitors (?avopiridol, paullones, hymenialdisine) constitutelead compounds with great potential for the treatment of AD and other ‘‘taupathies’’  Thebis-indole indirubin showspoor solubility, low absorption, and gastrointestinal toxicity. Several indirubin analoges, such as5-chloro-indirubin and indirubin-30-monoxime, have thus been synthesized for better pharmacological properties and reduced toxicity. Several mechanisms of action have been brought forward to explain the anti-mitotic and anti-tumoral properties of indirubins. Recently, thesecompounds were described as potent inhibitors of CDK’s by competing with ATP for binding to the catalytic site of the kinase.. It is con?rmed that indirubin inhibits GSK-3bas they do to CDKs by competing with ATP for binding to the catalytic site. The crystalstructure of CDK-2/indirubin-30-monoxime shows that the inhibitor binds, through three hydrogen bonds, to the backbone atoms of Glu-81 and Leu-83, two residues located in the ATP-binding pocket of the enzyme. The corresponding amino acids are Asp and Val in GSK-In general, good CDK’s inhibitors are good GSK-3 inhibitors, but there are two exceptions, 5,50-dibromo-indirubin ,which is quite active on GSK-3 but poorly active on CDK’s, and 5-SO3Na-indirubin.
Thebis-indole indirubin showspoor solubility, low absorption, and gastrointestinal toxicity. Several indirubin analoges, such as5-chloro-indirubin and indirubin-30-monoxime, have thus been synthesized for better pharmacological properties and reduced toxicity. Several mechanisms of action have been brought forward to explain the anti-mitotic and anti-tumoral properties of indirubins. Recently, thesecompounds were described as potent inhibitors of CDK’s by competing with ATP for binding to the catalytic site of the kinase.. It is con?rmed that indirubin inhibits GSK-3bas they do to CDKs by competing with ATP for binding to the catalytic site. The crystalstructure of CDK-2/indirubin-30-monoxime shows that the inhibitor binds, through three hydrogen bonds, to the backbone atoms of Glu-81 and Leu-83, two residues located in the ATP-binding pocket of the enzyme. The corresponding amino acids are Asp and Val in GSK-In general, good CDK’s inhibitors are good GSK-3 inhibitors, but there are two exceptions, 5,50-dibromo-indirubin ,which is quite active on GSK-3 but poorly active on CDK’s, and 5-SO3Na-indirubin.
C. Maleimides derivatives
Hymenialdisine
Concerning the family of maleimide-containing structures core, recent studies have demonstrated that the bisindolylmaleimide derivatives of staurosporine, GF 109203Âand Ro 31-8220, widely used as speci?c inhibitors of protein kinase C (PKC), are potent and direct inhibitors of GSK-3, was the more potent inhibitor,
MODE OF ACTION
The proposed mechanism of action is by competing with ATP for binding to the nucleotide-binding site of GSK-3b, such as it has been suggested for PKC. Recently, further biological effects has been described, ?nding that Ro 31-8220 in addition to inhibiting protein kinases also directly inhibits voltage-dependent
anilinomaleimide derivatives.
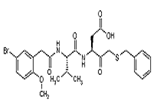
arylindolemaleimide family as potent GSK-3ainhibitors, showing equal potency for GSK-3b. In common with most of the other protein kinase inhibitors, these compounds inhibited GSK-3ain an ATP competitive manner. optimization process has been carried out in a variety of array formats via a combination of rational design,
Structure–activity relationship studies have de?ned some structural requirements for potency on this class of compounds. Concerning the substitutents in the aryl and aniline rings, the data suggest the greater aciditiy of phenol may result in the generation of stronger H-bond interaction or that potency is gained form additional lipophilicity, showing the best inhibitory results to compounds,Comparison among N-methylaniline16unsubstitued anilines ,and indoline derivative1 7(IC50¼161 nM) seems to point out that the conformational restraint of the indoline may allow a binding mode, which is unavailable to theN-methylaniline compounds, explaining the lower potency of these former compounds. Finally, low potency inhibitory of methylated compound (suggest that maleimide NH seems to be critical for the binding.As mentioned before, GSK-3 phosphorylates and modulates the activity of a number of key regulatory proteins in the cell. Pharmacological studies achieved in the maleimide derivatives, have showed that these compounds represent a valuable tool in elucidating the role of GSK-3bin cell signaling pathways. The treatment of cells with either compound stimulated responses characteristic of extracellular that are known to inhibit GSK- activity. SB-216763 and SB-415286 stimulate pathway.
MODE OF ACTION
Have shown the capability to protect primary neurones from death induced by reduced phosphatidylinositol 3-kinase pathway activity. The inhibition of neuronal death correlated with inhibition of GSK-3 activity and modulation of GSK-3 substrates and b-catenin. Following with the neuroprotective effect resulting from GSK-3 inhibition, recent evidences indicate that adenoviral FRAT1 overexpression in mammalian cells is neuroprotective and that neuroprotection correlates with inhibition of GSK-3 activity towards tau and b-catetin. By comparison, treatment with SB-216763 and SB-415286 (2 0) was found to be more potent than FRAT1 overexpression in terms of neuroprotection, suggesting the therapeutic value of these compounds in the treatment of neurodegenerative disorders such as Alzheimer’s disease or for the prevention of neuronal apoptosis after a stroke.
d. MuscarinicAgonist
Recently cholinergic compounds such as M1 muscarinic agonist have shown potential not only as cholinergic drugs for cognition enhacement, but also as disease-modifying agents in AD promoting the nonamyloidogenic amyloid protein precursor (APP) processing pathways and decreasing tau protein hyperphosphorylation.67Some of them, including AF102B (21) and AF150 (2 2) reduce tau phosphorylation via GSK-3 inhibition.68This unique property of M1 agonist to alter different aspects of AD pathogenesis could represent the most remarkable, yet unexplored, clinical value of such compounds.
e.ATP noncompetitive inhibitor.
Thiadiazolidinones
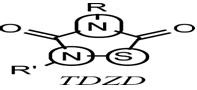
The 2,4-disubstituted thiadiazolidinones (TDZD) are described as the first ATP-noncompetitive GSK-3 inhibitors. Following an SAR study about TDZD, different structural modifications in the heterocyclic ring aimed to test the influence of each heteroatom on the biological study are here reported here. Various compounds such as hydantoins, dithiazolidindiones, rhodanines, maleimides, and triazoles were synthesized and screened as GSK-3 inhibitors. After an extensive SAR study among these different heterocyclic families, TDZDs have been revealed as a privileged scaffold for the selective inhibition of GSK-3. several therapeutic uses, including the treatment of neurodegenerative diseases, bipolar disorders, stroke, cancer, and chronic in?ammatory disease. As there is lot of recent literature dealing with the involvement of GSK-3 in the molecular pathways of different diseases, this review is mainly focused on the new GSK-3 inhibitors discovered or speci?cally developed for this enzyme, their chemical structure, synthesis, and structure–activity relationships, with the aim to provide some clues for the future optimization of these promising drugs.Small thiadiazolidinones (TDZD) derivatives are the ?rst ATP-non competitive GSK-3 inhibitors reported to date.The inhibitory kinase activity of these compounds, considering as secondary products in the obstention of potassium channel openers,muscarinic agonist,or acetylcholinesterase inhibitors,73was discovered in a GSK-3 directed screening program initiated two years ago. Thiadiazolidinones (23) were synthesized following a pathway which is based on the reactivity Of N-alkyl-S-[N0-(chlorocarbonyl)amino]isothiocarbamoyl chlorides with isocyanates.74Currently, their structural key features have been established.
structure–activity relationship studies point out to the size and the nature of the substituents attached to both nitrogen atoms of the thiadiazolidinone as crucial features for GSK-3 inhibition. Additionally, carbonyl or thiocarbonyl moieties in a 1,3-disposition on the heterocyclic framework may be critical for binding. Moreover, a hypothetical GSK-3 binding mode has been proposed in where the TDZD derivatives may bind to the primed phosphate substrate binding site of the kinase. Interactions of TDZD with the main amino acids involved in this recognition site (Arg 96, Lys 205, Tyr 216) of GSK-3 explain the structure–activity relationships found in this family of compounds until now. TDZD do not show inhibition on others several kinases as PKA, PKC, CK-2, and CDK1/cyclin B, and studies in primary culture neuronal cells have demonstrated that tau phosphorylation decrease in the presence of these inhibitors.These data point to promising drugs for the treatment of Alzheimer’s disease,but further development must assess their clinical use .(21,22,23,24,25,26)
CONCLUSION & SUMMARY
GSK-3 has a central role in several accute and their inhibitors arises as promising drugs for their pharmacotherapy. It is worth mentioning that GSK-3 is investigated as a candidate gene for AD77being the human GSK-3bpromoter isolated.Additionally, its inhibitors and antisenseoligonucleotides exert protection against neuronal death.79Small molecules inhibitors of GSK-3 may, therefore, have several therapeutic uses, including the treatment of neurodegenerative bipolar disorders, stroke, and in?ammatory disease. However, there are some possible obstacles for these promising drugs. In the heart, GSK-3 appears to suppress cardiac hypertrophy.Chronic suppression of this kinase may, therefore, lead to cardiac complications.
Further, the GSK-3 blockers increase cellularb-catenin levels, which may increase the propensity for developing certain cancers.Herein lies the potential problem with the GSK-3 inhibitors that have been developed so far.
These compounds are ATP competitive, and therefore, block the phosphorylation of every substrate, not just those involved in insulin signaling or tau phosphory- lation. However, the discovery of the way in which ‘‘primed’’substrates dock with GSK-3 might have opened a new opportunity to develop compounds that target only the site that binds the ‘‘priming phosphate’’ of substrates. So, TDZD compounds, the only non ATP-competitive GSK-3 inhibitors known until the moment, might be less likely to be oncogenic in contrast to ATP- competitive inhibitors. Clearly, the clinical utility of these drugs awaits animal and human trials.
SCOPE OF STUDY
• .This study gives all information about Glycogen synthase kinase and their use in CNS therapies.
• This study also has information about drug present today and its chemical structure site of action and its side effect and whether this drug gives the effect present toward disease condition such as Alzheimer’s disease
• This study include a recent information about drug till present and are under research
• This study can provide suitable information to those which are working on Glycogen synthase kinase inhibitor and for its development and its modification
• This study can make work easy for research and development of Glycogen synthse kinase inhibitor by providing the information about site of action of drug.
An attempt to include various approaches Glycogen synthase kinase inhibitor has been include in this project.
REFERENCE
1. Woodgett JR (August 1994). "Regulation and functions of the glycogen synthase kinase-3 subfamily". Cancer Biol. 5 (4): 269–75. PMID 7803763.
2. Woodgett JR (September 2001). "Judging a protein by more than its name: GSK-3". Sci. STKE 2001 (100): RE12. doi:10.1126/stke.2001.100.re12. PMID 11579232.
3. Ali A, Hoeflich KP, Woodgett JR (August 2001). "Glycogen synthase kinase-3: properties, functions, and regulation". Chem. Rev. 101 (8): 2527–40. doi:10.1021/cr000110o. PMID 11749387.
4. Cohen, P. (2002) Protein kinases – the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315
5. 5.Noble, M.E. et al. (2004) Protein kinase inhibitors: insights into drug design from structure. Science 303, 1800–1805
6. sciencedirect.com 0165-6147/$ - see front matter Q 2004 Elsevier Ltd. All rights reserved. doi:10.1016/j.tips.2004.07.006
7. Fujimuro, M. et al. (2003) A novel viral mechanism for dysregulation of beta-catenin .
8. Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 9, 300–306
9. Davies, S.P. et al. (2000) Specificity and mechanism of action of somen commonly used protein kinase inhibitors. Biochem. J. 351, 95–105
10. Bain, J. et al. (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204
11. Jackson, G.R. et al. (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34, 509–519
12. Noble, W. et al. (2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38, 555–565
13. Bhat, R.V. et al. (2000) Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U. S. A. 97, 11074–11079
14. Wang, J. et al. (2003) Cdk5 activation induces hippocampal CA1 cell death byby directly phosphorylating NMDA receptors. Nat. Neurosci. 6, 1039–1047
15. R. V. Bhat et al..Alzheimer’s disease, bipolar disorder, phosphorylation, schizophrenia, signal transduction, Tau kinase, ternational Society Infor Neurochemistry ,J. Neurochem. (2004) 89, 1313–1317.
16. ncbi.nlm.nih.gov/pubmed/15189333
17. scribd.com/doc/38725570
18. Klein PS, Melton DA. Proc Natl Acad Sci USA. 1996;93:8455–8459.
19. ncbi.nlm.nih.gov/pubmed/15559249
20. Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. Neuropsychopharmacol. 2004;29:142PMC1850891/
21. freepatentsonline.com/7098204.
22. ncbi.nlm.nih.gov/pmc/articles/PMC1850891
23. google.co.in/imgres?imgurl=http://structuresearch.merck-chemicals.com/getImage/EMD_BIO_12687
24. circ.ahajournals.org/cgi/content/full/circulationaha;109/10/1196
25. google.co.in/imgres?imgurl
26. bindingdb.org/data/jpeg/tenK0/BindingDB_5655
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








