

About Authors
Ms. Priti B. Savant,Ms. Ashwini R. Pawar, Ms. Kaufiya D. Sayyed , Ms. Pooja R. Yelmar
1,2,3,4 Sahyadri College of Pharmacy Methwade Tal, Sangola, Distsolapur Maharashtra 413307
ABSTRACT :
Computer-aided drug design (CADD) provides a variety of tools and techniques that assist in the various stages of drug design, thereby reducing the cost of drug research and development time. Drug discovery and the development of a new drug is a long, complex, costly and highly risky process that has no equal in the commercial world. Therefore, computer-aided drug design (CADD) approaches are widely used in the pharmaceutical industry to speed up the process. The cost advantage of using computational tools in the lead optimization phase of drug development is significant. The cost and time invested by pharmacological research laboratories are heavy at various stages of drug discovery, starting from therapeutic target setting candidate drug discovery to evaluating the efficacy and safety of newly developed drugs, drug optimization through preclinical and extensive clinical trials. Major pharmaceutical companies have invested heavily in routine ultra High Throughput Screening (uHTS) of large numbers of drug-like molecules. In parallel, drug design and optimization are increasingly using computers for virtual screening. Recent advances in DNA microarray experiments are discovering that thousands of genes involved in a disease can be used to gain in-depth information about disease targets, metabolic pathways, and toxicity of drugs. Theoretical tools include empirical molecular mechanics, quantum mechanics, and more recently statistical mechanics. This latest advance allowed the inclusion of overt solvent effects. All this is largely the availability of high-quality computer graphics supported on workstations.
INTRODUCTION :
Computer-aided drug design (CADD) provides a variety of tools and techniques that assist in the various stages of drug design, thereby reducing the cost of drug research and development time. Drug discovery and the development of a new drug is a long, complex, costly and highly risky process that has no equal in the commercial world. Therefore, computer-aided drug design (CADD) approaches are widely used in the pharmaceutical industry to speed up the process. The cost advantage of using computational tools in the lead optimization phase of drug development is significant. The cost and time invested by pharmacological research laboratories are heavy at various stages of drug discovery, starting from therapeutic target setting candidate drug discovery to evaluating the efficacy and safety of newly developed drugs, drug optimization through preclinical and extensive clinical trials. Major pharmaceutical companies have invested heavily in routine ultra High Throughput Screening (uHTS) of large numbers of drug-like molecules. In parallel, drug design and optimization are increasingly using computers for virtual screening. Recent advances in DNA microarray experiments are discovering that thousands of genes involved in a disease can be used to gain in-depth information about disease targets, metabolic pathways, and toxicity of drugs. Theoretical tools include empirical molecular mechanics, quantum mechanics, and more recently statistical mechanics. This latest advance allowed the inclusion of overt solvent effects. All this is largely the availability of high-quality computer graphics supported on workstations.
|
Sr,No.
|
DRUG |
DISEASE |
TARGET |
|
1 |
Oxymorphone |
Opioid analgesic |
Agonist of mu-opiniod receptor |
|
2 |
Saquinavir |
AIDS |
Inhibits proteases of HIV1 and HIV 2 |
|
3 |
Captopril |
Hypertension or high BP |
Inhibits conversion of angiotensin-converting enzyme |
|
4 |
Zanamivir |
Affects influenza A and influenza B |
Inhibits neuraminidase |
|
5 |
Dorzolamide |
Glaucoma |
Inhibits carbonic anhydrase |
DRUG DISCOVERY AND DEVELOPMENT PROCESS
Drug discovery can be defined as the process of identifying chemical entities that have the potential to be therapeutic agents. An important role of drug discovery campaigns is the recognition of new molecular entities that may be valuable in the treatment of diseases characterized as unmet medical needs. The development of a drug is a very complex process that can take about 5-10 years from the first idea to hitting the market and cost USD 1.7 billion. A new development idea, current requirements of the market, emerging diseases, academic and clinical research, commercial sector, etc. It can come from a variety of sources, including Once a target has been selected for discovery, the pharmaceutical industries or related academic centers work on early processes to identify chemical molecules with suitable properties to make targeted drugs.
In the last few years, CADD has grown rapidly, reinforcing the perception of multifaceted and difficult biological processes. With the help of these computational tools, it is now possible to find new pharmacologically active agents in a short time.
HISTORY OF CADD :
A Brief History of CADD In 1900, the concept of receiver and lock-key was given by P.Ehrich (1909) and E. Fisher. In the 1970s the concept of Quantitative structure-activity relationships (QS-AR) was established, It had limitations: 2-Dimensional, retrospective analysis; In the 1980s, CADD Molecular Biology, X-ray crystallography, multidimensional NMR along with computer graphics started the era of molecular modeling. In the 1990s, more modern techniques such as Human genome Bioinformatics were introduced in the Innovative world of medical science along with Combinatorial chemistry and High throughput screening.
DRUG DISCOVERY PROCESS :
From the original idea to the launch of a finished product, developing a new drug is a complex process that can take 12-15 years and costs more than USD 1 billion. A goal idea can come from a variety of sources, including academic and clinical research, and the commercial sector. It may take many years to generate supporting evidence before choosing a target for an expensive drug discovery program. Once a target has been selected, the pharmaceutical industry and, more recently, some academic centers streamlined a series of early processes to identify molecules with properties suitable for making acceptable drugs. Product Characterization.
• Formulation, Delivery, Packaging Development.
• Pharmacokinetics and Drug Disposal.
• Preclinical Toxicology Test and IND Application.
• Bioanalytical Test.
• Clinical trials.
Various stages of drug design
• Choose a disease
• Choose a drug target
• Define a bioassay
• Find a precursor compound
• Isolate and purify lead compound if necessary
• Determine the structure of the lead compound
• Define the structure Activity relationship
• Identify the pharmacophore
• Improve target interaction.
Commonly Used Methods in Drug Design
Drug Design can basically be divided into two types: Ligand-based drug design (LBDD) and Structure-based drug design (SBDD)
• Ligand-based drug design or indirect drug design
• Structure-based drug design or direct drug design
• Rational drug design
• Computer aided drug design
1] Ligand-based drug design or indirect drug design
Ligand-based drug design is an approach used in the absence of receptor 3D information and relies on knowledge of molecules that bind to the biological target of interest. 3D quantitative structure activity relationships (3D QSAR) and pharmacophore modeling are the most important and widely used tools in ligand-based drug design. They can provide appropriate predictive models for lead identification and optimization.
Ligand-based drug design is an approach used in the absence of receptor 3D information and relies on knowledge of molecules that bind to the biological target of interest.
a) Ligand-Based Drug Design consists of the information of molecules that bind to the desired target site.
b) These molecules can be used to derive a Pharmacophore model.
c) A pharmacophore model is defined as a molecule with the necessary structural abilities to bind to a desired target site.
d) Once the Pharmacophore is identified, it is determined whether it is suitable for the receptor, otherwise the Pharmacophore is further modified to make a potential drug.[4]
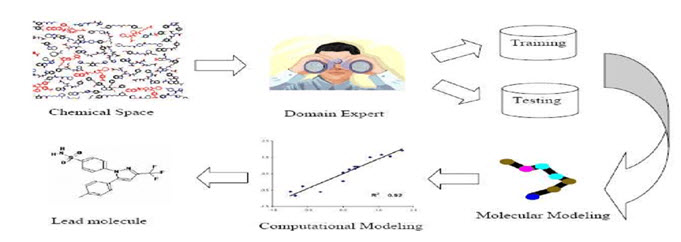
Figure-1 : General steps involved in ligand based drug design
Important tools used in ligand-based drug design
1] Quantitative structure-activity relationships (QSAR)
Quantitative structure-activity relationship models are regression or classification models used in the chemical and biological sciences and engineering. Like other regression models, QSAR regression models correlate a number of "predictive" variables with potency.
response variable (Y), while the classification QSAR models correlate the estimator variables are converted to a categorical value of the response variable. In order to place 10 different groups at the 4 positions of the benzene ring, the number of compounds required for synthesis is 10. Solution: Synthesize a small number of compounds and derive from their data.rules for predicting the biological activity of other compounds.
A] VEGA platform [https://www.vegahub.eu/portfolio-item/vega-qsar/]
Using the VEGA platform, you can access a range of QSAR models for regulatory purposes or develop your own for research purposes. QSAR models can be used to predict the property of a chemical compound using information from its structure.
B] DEMETRA [http://www.demetra-tox.net/]
DEMETRA is an EU funded project. The aim of this project was to develop predictive models and software that give a quantitative estimate of the toxicity of a molecule, specifically pesticide molecules, candidate pesticides and their derivatives. The input is the chemical structure of the compound.
software algorithms use “Quantitative Structure-Activity Relationships” (QSARs). The DEMETRA software tool can be used for toxicity estimation of pesticide molecules and related compounds. DEMETRA
Models are freely available. Five models were developed to predict toxicity to trout, daphnia, quail and bees. The software is based on the homogeneous integration of the knowledge acquired in the DEMETRA EU project, using the best algorithms obtained as the basis for hybrid combination models to be used for prediction purposes.
C] T.E.S.T [https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test]
The Toxicity Estimation Software Tool (T.E.S.T.) will allow users to easily estimate acute toxicity using the above QSAR methodologies.
D] OCHEM [https://ochem.eu/home/show.do]
OCHEM is an online database of experimental measurements integrated with the modeling environment. Submit your experimental data or use data uploaded by other users to create predictive QSAR models for physical-chemical or biological properties.
E] E-DRAGON [http://www.vcclab.org/lab/edragon/]
E-DRAGON by Milan Chemometrics and QSAR Research Group by Prof. It is an electronic remote version of the well-known software DRAGON, an application for the calculation of molecular descriptors developed by R. Todeschini. These descriptors can be used for molecular structure-activity or structure-property relationships as well as similarity analysis and high-throughput screening of the molecule database.
F] SeeSAR [https://www.biosolveit.de/SeeSAR/]
SeeSAR is a software tool for interactive, visual composite prioritization and composite evolution. Structure-based design work ideally supports a multi-parameter optimization to maximize the probability of success rather than similarity alone. One of SeeSAR's strengths is that the relevant parameters are at hand, along with real-time visual computer assistance in 3D.
G] Dragon [https://chm.kode-solutions.net/products_dragon.php]
Dragon calculates 5,270 molecular descriptors covering most of the various theoretical approaches. The list of descriptors includes the simplest types of atoms, functional groups and part counts, topological and geometric descriptors, three-dimensional descriptors, as well as several property predictions (such as logP) and drug-like and lead-like stimuli (such as Lipinski's). Alarm).
H] PaDEL-Descriptor [http://www.yapcwsoft.com/dd/padeldescriptor/]
A software to calculate molecular identifiers and fingerprints. The software currently calculates 1875 identifiers (1444 1D, 2D identifiers and 431 3D identifiers) and 12 types of fingerprints (16092 bits total).
Identifiers and fingerprints are calculated using The Chemistry Development Kit, which has additional identifiers and fingerprints such as atom type electrotopological state descriptors, Crippen logP and MR, extended topochemical atom (ETA) descriptors, McGowan volume, molecular linear free energy relationship descriptors, ring numbers. The number of chemical substructures and binary fingerprints identified by Laggner and the number of chemical substructures identified by Klekota and Roth.
2] PHARMACOPHOR
Molecular similarity-based search is the simplest LBDD technique to identify desired small molecules. Molecular similarity-based searching is both an independent and integral part of other LBDD and SBDD techniques, where small molecule libraries are searched using molecular identifiers. Molecular descriptors are characteristic numerical values that represent small molecules and range from simple physicochemical properties to complex structural properties. Examples of molecular descriptors include molecular weight, atom types, bond distances, surface area, electro-negativities, atomic distributions, aromaticity indices, solvent properties, and others. Molecular descriptors are derived through experiments, quantum-mechanical tools, or prior knowledge. Depending on the "dimensionality", molecular identifiers can be 1D, 2D or 3D identifiers. 1D descriptors are scalar physicochemical properties of a molecule, such as molecular weight, logP values, and molar refraction. 2D identifiers are derived from molecular structure or configuration and include topological indexes and 2D fingerprints. 3D descriptors are derived from the conformation of molecules. 3D descriptors can identify 3D fingerprints, dipole moments, highest occupied molecular orbital/lowest unoccupied molecular orbital energies, electrostatic potentials, etc. includes. A list of software that predicts molecular annotation
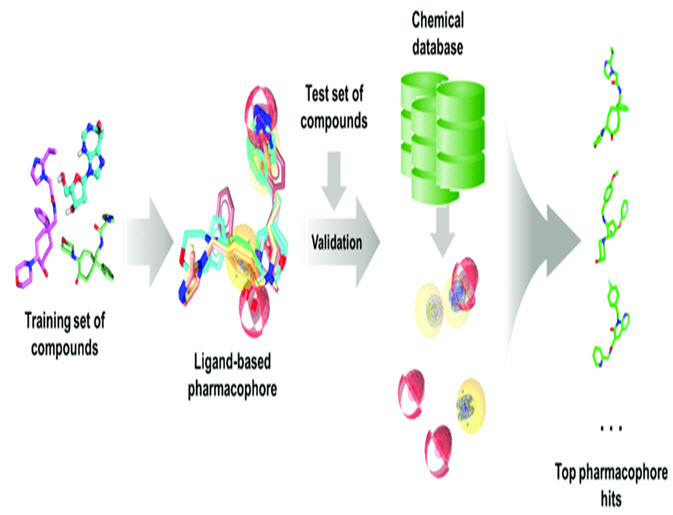
Figure-2 : Typical ligand based pharmacophore generation and screening workflow
Table-1 : Common software to predict molecular descriptors
|
Software |
Number of Types of Prediction Detectors
|
|
ADAPT |
>260 (topological, geometrical, electronic, physicochemical)
|
|
ADMET Predictor |
>290 (constitutional, functional group counts, topological, E-state, moriguchi descriptors, meylan flags, molecular patterns, electronic properties, 3D descriptors, hydrogen bonding, acidebase ionization, empirical estimates of quantum descriptors) |
|
CODESSA
|
>1500 (constitutional, topological, geometrical, charge related, semiempirical, thermodynamical) |
|
DRAGON |
>5200 (constitutional, topological, 2D autocorrelations, geometrical, WHIM, GETAWAY, RDF, functional groups, properties, 2D binary and 2D frequency fingerprints, etc.) |
|
MARVIN Beans |
>500 (physicochemical, topological, geometrical, fingerprints, etc.)
|
|
MOE |
>300 (topological, physical properties, structural keys, etc.)
|
|
MOLGENQSPR |
>700 (constitutional, topological, geometrical, etc.)
|
|
PreADMET |
>955 (constitutional, topological, geometrical, physicochemical, etc.)
|
2] Structure-based drug design or direct drug design
Structure-based drug design (SBDD) is the process that includes virtual screening and de novo drug design. These methods are a highly efficient and alternative approach to the discovery and development of the drug design course. In virtual scanning, drug chemical compounds are computationally screened against known target structure [5,6]. In classical or advanced pharmacology or legacy drug design and development, rational drug design is very costly and efficient. The first step in rational drug design method or reverse pharmacology is to identify promising target proteins used for screening small molecule libraries. Structure-based virtual scanning (SBVS), molecular docking and molecular dynamics (MD) are methods used in SBDD, a more specific, efficient and rapid process for lead discovery and optimization, because they are approximately related to the 3D structure of a Target protein. analysis of disease and binding energies at the molecular level, ligand protein interaction induction insertion process.[5,6] There are many drugs identified by SBDD with the help of some techniques such as thymidylate synthase inhibitor, raltitrexed and potential HIV protease inhibitor. these were discovered by MD simulation and the antibiotic norfloxacin [7,8,9,10]. The three-dimensional (3D) structure of proteins (more than 100,000) is provided in SBDD.

Figure-3 : Workflow of Structure Based Drug Design
The various principle and efficient methods for SBDD workflow

1] Identification of target protein and binding site:
Target protein identification is the key step in the SBDD process. It provided clear information on the binding site of the target macromolecule, protein-ligand interaction, post-docking dynamics, as well as hydrogen bond formation, which helped to calculate the best pharmacophores of the 'new' ligand. The binding sites determined experimentally by integrative structural biology techniques in the 3D structure of the target macromolecule such as NMR, X-ray crystallography. The next step is to identify the binding pocket after the target protein is resolved. It is a very small space where the ligand binds and also exerts its therapeutic or desired effect. These methods provide information on energy interaction and Van der Waals (vdW) forces for binding site mapping. There are many methods developed by energy interaction calculations for binding site mapping specifically for SBDD, and these methods identify specific regions of the target protein that interact with appropriate functional groups on drugs. These identify with the protein Q-site Finder [11][12][13][14]
2] Molecular docking
Molecular docking is a virtual simulation technique used to model the interaction between a small molecule and a protein at the atomic level. This technique is also used to characterize the behavior of small molecules at the binding site of the target protein The insertion process involves two basic steps - the estimation of ligand conformation and the second is the binding of the ligand within the target active site with accuracy, so this technique is widely used in structure-based drug design (SBDD).
3] Scoring function
The scoring function assists an insertion program into the ligand binding site. The scoring function also helps calculate the binding affinity between protein and ligand functions. Scoring functions are divided into force field, empirical, knowledge-based, and machine learning.
An early general-purpose empirical scoring function was developed by Bohm to describe the binding energy of ligands to receptors
Table-2 : Softwares for Structure-Based Drug Designing (SBDD)
|
Stages |
Tools used |
Brief Description |
Links |
|
1.Target modeling |
SWISS-MODEL |
Homology modeling |
https://swissmodel.expasy.org/ |
|
|
MODELER |
Homology Modelling |
https://salilab.org/modeller/ |
|
|
Phyre and Phyre2 |
Template detection alignment as well as 3D modeling |
http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index |
|
2.Binding site |
CASTp |
Binding site prediction |
http://sts.bioe.uic.edu/castp/index.html?201l |
|
|
Active site prediction tool |
Active site prediction |
http://www.scfbio-iitd.res.in/dock/ActiveSite.jsp |
|
3. Molecular Docking |
AutoDockVina |
Molecular docking and virtual screening |
https://vina.scripps.edu/ |
|
|
Schrodinger |
Maestro |
https://www.schrodinger.com/products/maestro
|
CADD in the drug discovery process
CADD can be combined with wet laboratory techniques to elucidate and accelerate the drug discovery process to design new drugs (eg antibiotics) for both known and novel targets. CADD simplifies the drug design process by minimizing time and cost.[15[16][17]
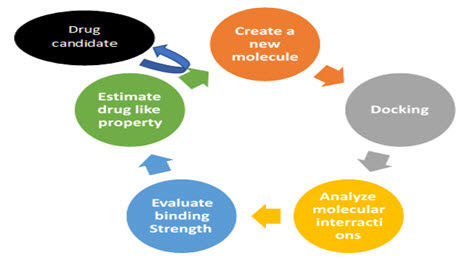
Figure4 : CADD Process
Advantages of CADD
1. A cost-effective, time-saving, fast and automated process.
2.It give an idea about the drug-receptor interaction pattern.
3. Minimize synthetic and biological testing efforts.
4. Minimize the possibility of failure in the final stage.
REFERENCES :
1. A review on computer aided drug design,international journal of emerging technologies and innovatives research page no.1165-1176 march 2020.
2. Song CM, Lim SJ, Tong JC. Recent advance in computer aided drug design. Brief in bio information. 2009.
3. Hughes, J P et al. “Principles of early drug discovery.” British journal of pharmacology vol. 162,6 (2011): 1239-49. doi:10.1111/j.1476-5381.2010.01127.x
4. Crasto AM. All About Drugs. Mumbai, India:[Publisher unknown]; Available from: http://www.allfordrugs.com/drug-design/
5. Lionta E., Spyrou G., Vassilatis D.K., Cournia Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014;14:1923–1938. doi: 10.2174/1568026614666140929124445.
6. Kalyaanamoorthy S., Chen Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today. 2011;16:831–839. doi: 10.1016/j.drudis.2011.07.006.
7. Anderson A.C. The process of structure-based drug design. Chem. Biol. 2003;10:787–797. doi: 10.1016/j.chembiol.2003.09.002.
8. Wlodawer A., Vondrasek J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu Rev. BiophysBiomol. Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249.
9. 20. Clark D.E. What has computer-aided molecular design ever done for drug discovery? Expert Opin. Drug Discov. 2006;1:103–110. doi: 10.1517/17460441.1.2.103.
10. 21. Rutenber E.E., Stroud R.M. Binding of the anticancer drug zd1694 to E. Coli thymidylate synthase: Assessing specificity and affinity. Structure. 1996;4:1317–1324. doi: 10.1016/S0969-2126(96)00139- 6.
11. Laurie A.T., Jackson R.M. Q-sitefinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916.
12. Zhang Y.; hand.; Tian H.; Jiao Y.; Shi Z.; Ran T.; Liu H.; Lu S.; Xu A.; Qiao X.; Pau J.; Yin L.; Zhou W.; Lu T.; Chen Y.; identification of covalent binding sites targeting cryteines based on computational approaches Mol. Pharma,2016,13(9) 3106-3118.
13. Grant M.A. Protein structure prediction in structure-based ligand design and virtual screening. Comb. Chem. High Throughput Screen. 2009;12:940–960. doi: 10.2174/138620709789824718.
14. Pau L.; Gardner, C.L.; Pugliai, F.A.; honzalez, teleonomic acid binding pocket in prb from liberibacterasiaticus. Front microbiol.,2017,8,1591.
15. Suchitra Ajjarapu;Computer-Aided Drug Design (CADD)- Definition, Types, Uses, Examples, SoftwaresFebruary 25, 2022 https://thebiologynotes.com/computer-aided-drug-design-cadd/.
16. Tardif JC, L'allierPL, Ibrahim R, Grégoire JC, Nozza A, Cossette M, Kouz S, Lavoie MA, Paquin J, Brotz TM, Taub R, Pressacco J. Treatment With 5-Lipoxygenase Inhibitor VIA-2291 (Atreleuton) In Patients With Recent Acute Coronary Syndrome. Circ. Cardiovasc. Imaging. 2010;3(3):298–307
17. Janvi Gajipara1 And John J Georrge; TOOLS FOR LIGAND BASED DRUG DISCOVERY ; Recent Trends In Science And Technology-2018 (February 11, 2018)








