
{ DOWNLOAD AS PDF }
 About Authors:
About Authors:
Mr. Pranab Prakash Panigrahi1*, Mr. Ajit Kumar Acharya2
1B.Pharm, ROYAL COLLEGE OF PHARMACY AND HEALTH SCIENCES, BERHAMPUR
2Asst.Professor, ROYAL COLLEGE OF PHARMACY AND HEALTH SCIENCES, BERHAMPUR
*pranab.panigrahi@rediffmail.com
INTRODUCTION
Poorly water-soluble drugs often require high doses in order to reach therapeutic plasma concentrations after oral administration. Improvement in the extent and rate of dissolution is highly desirable for such compounds, as this can lead to an increased and more re-producible oral bioavailability and subsequently to clinically relevant dose reduction and more reliable therapy. More than 40% of newly discovered drugs have little or no water solubility presents a serious challenge to the successful development and commercialization of new drugs in the pharmaceutical industry. Now a days, pharmaceutical technology provides many approaches to enhance the dissolution rate of poorly soluble drugs. Physical modifications often aim to increase the surface area, solubility and/or wet ability of the powder particles and are therefore focused on particle size reduction or generation of amorphous states [Hancock, 1997 & Grau, 2000]. Several methods have been employed to improve the solubility of poorly water soluble drugs. A solid dispersion technique has been used by various researchers who have reported encouraging results with different drugs. The first drug whose rate and extent of absorption was significantly enhanced using the solid dispersion technique was sulfathiazole by Sekiguchi and Obi (Sekiguchi, 1961)1.
REFERENCE ID: PHARMATUTOR-ART-1475
Based on their solubility and intestinal membrane permeability characteristics, drug substances have been classified into one of four categories according to the BCS (Fig. 1). The BCS is one of the most significant prognostic tools created to facilitate oral drug product development in recent years; the validity and broad applicability of the BCS have been the subject of extensive research and discussion; it has been adopted by the US Food and Drug Administration (FDA), the European Medicines Agency (EMEA), and the World Health Organization (WHO) for setting bioavailability/bioequivalence (BA/BE) standards for immediate-release (IR) oral drug product approval; and the BCS principles are extensively used by the pharmaceutical industry throughout drug discovery and development 2.
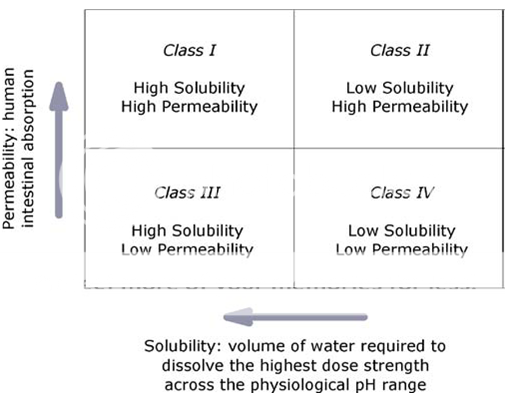
Fig. 1.The BCS classifies drugs by their solubility and permeability properties in order to stand for the most fundamental view of the drug intestinal absorption process following oral administration.
Formulation of poorly water-soluble drugs in the most stable dosage form for oral delivery perhaps presents the greatest challenge to pharmaceutical industry. Physical transformation of drug substance into its more soluble but metastable amorphous form is one of the approaches for improving dissolution rate of such drugs. The present study utilizes technique of spray drying for preparation of solid dispersions (SDs) and includes stability study of the same. Valdecoxib (VLD), a prototype of poorly water-soluble drugs, has been the drug of choice3.
[adsense:468x15:2204050025]
In an attempt to prepare a new water-soluble, parenteral COX-2 inhibitor, Rofecoxib and Celecoxib analogues were designed and synthesized for evaluation as selective cyclooxygenase-2 (COX-2) inhibitors with in vivo anti-inflammatory activity. In this experiment, respective SO2Me and SO2NH2 hydrogen-bonding pharmacophores were replaced by a tetrazole ring. Molecular modeling (docking) studies showed that the tetrazole ring of these two analogues was inserted deep into the secondary pocket of the human COX-2 binding site 4.
Intermolecular interactions and the ability of the solvent to form a hydrogen bond with the drug molecules were found to be the major factors involved in the dissolution of drugs by pure solvents. There are various methods like freeze drying, physical mixing, fusion (melt) method, solvent evaporation etc. are employed for the formulation of solid dispersion and Inclusion complexes by various methodologies i.e. co-precipitation/co-evaporation, kneading, Complexation, micellar solubilisation etc. to enhance its aqueous solubility and in-vitro dissolution rate.
Solubility is defined as “ability of a substance to form a solution with another substance” Intrinsic solubility is defined as the maximum concentration to which a solution can be prepared with a specific solute and solvent.
Solubility means when a solid is placed in contact with solvent, molecules or ions detach themselves from the surface of the solid and diffuse throughout the solvent, these ions or molecules therefore become molecularly dispersed. However some of the solute may return to the surface of the solid and therefore pass back out of solution. The process will continue until the solubility limit of the material is exceeded, at which point equilibrium is reached between the dissolved and the undissolved material. The resulting solution is saturated solution. Saturated solutions are normally prepared by adding an excess of solute to a solvent and applying heat. The prepared solution is then cooled to room temperature in the presence of undissolved material.
An understanding of the solubility of drug can be regarded as the most important aspect of pre-formulation testing.
The term solubilization is generally applied to the solution of predominantly lipophilic molecule in an aqueous surfactant solution; it may be equally well applied to the solution of non-aqueous surfactant solution, which contains reverse micelles.
Dissolution is defined as the maximum amount of solid that goes into solution under standard conditions of temperature, pH and solvent composition.
Statements of solubility are indicated by a descriptive phrase and are intended to apply at 20-300C. The following table indicates the meaning of the terms used in statements of approximate solubility5.
|
Descriptive Term |
Approximate volume of solvent in milliliter per gram(ml/gm) of solvent |
|
Very Soluble |
Less than 1part |
|
Freely Soluble |
1 to 10 parts |
|
Soluble |
10 to 30 parts |
|
Sparingly soluble |
30 to 100 parts |
|
Very insoluble |
1000 to 10,000 parts |
|
Insoluble` |
More than 10,000 parts. |
The term poorly soluble is used to describe a mixture of which only some of the component dissolves.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
IMPORTANCE OF SOLUBILITY:
* It helps for the identification of potential screening and bioavailability issues. It is important for the confirmation of bioavailability issues.
* It is important for the confirmation of bioavailability issues. During early trials of drugs it is used in the design of animal formulations as well as human formulation design.
* Solubility knowledge is needed for biopharmaceutical classification, biowaivers, and bio equivalence. It is also required for formulation optimization and salt selection. Solubility also effects the optimization of manufacturing process.
REASON FOR INSOLUBILITY OF A COMPOUND:
The solubility of a solute in a solvent is equal to the thermodynamic activity of the solute, usually in the pure state, divided by the activity coefficient of the solute in the saturated solution. The thermodynamic activity of the pure solute measures the escaping tendency of the solute from the pure state and decreases with increasing strengths of the solute-solute interactions, which for solid, is reflected in the melting point and lattice energy. Modification of solid state such as the formation of amorphous forms, metastable polymorphs, co-crystals, or novel particles, cans abolish or lower the lattice energy and can therefore increase the solubility of a compound.
PROCESS OF SOLUBILISATION: 6
The process of solubilisation involves the breaking of inter-ionic or intermolecular bonds in the solute, the separation of the molecules of the solvent to provide space in the solvent for the solute, interaction between the solvent and the solute molecule or ion.
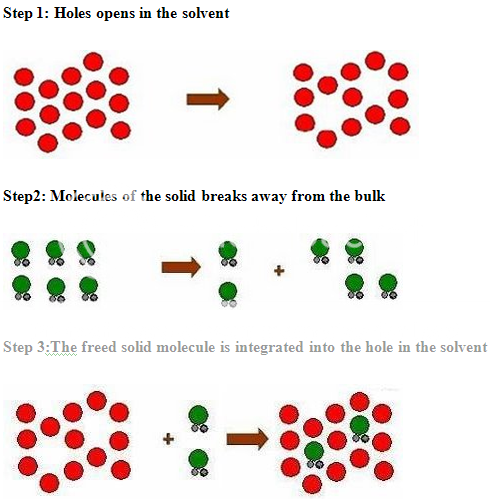
The various formulation and chemical approaches that can be taken to improve the solubility or to increase the available surface are for dissolution are as given below:
1) Physical modification
a) Particle size
· Micronization
· Nanosuspensions
b) Modification of crystal habit
· Polyomorphs
· Pseudopolyomorphs (including solvates/hydrates)
c) Complexation/Solubilization
· Use of co-solvents
· Use of surfactants
· Use of cyclodextrins
d) Drug dispersion in carriers
· Eutectic mixtures
· Solid dispersion (non-molecular)
2) Chemical modification
· Soluble prodrugs
· Salts
LITERATURE REVIEW
1. Bada Pragati Kumar et al. studied about Efavirenz is an HIV-1 specific, non-nucleoside reverse transcriptase inhibitor (NNRTI).It is an antiretroviral agent indicated for the treatment of human Immunodeficiency virus type 1 (HIV-1) infection, which is not soluble in water and lower absorption in gastric fluid.
2. Arik Dahan et al. explained the Biopharmaceutics Classification System (BCS) categorizes drugs into one of four biopharmaceutical classes according to their water solubility and membrane permeability characteristics and broadly allows the prediction of the rate-limiting step in the intestinal absorption process following oral administration.
3. Anshuman A et al. viewed stability study of amorphous valdecoxib. Physical transformation of drug substance into its more soluble but metastable amorphous form is one of the approaches for improving dissolution rate of such drugs.
4. Latifeh Navidpour et al discussed the design and synthesis of new water-soluble tetrazolide derivativesof celecoxib and rofecoxib as selective cyclooxygenase-2(COX-2) inhibitors.
5. C.A. Ventura et al. evaluated the influence of modified cyclodextrins {hydroxypropyl--cyclodextrin (HP-Cyd) and 2, 6-di-O-methyl-cyclodextrin (DM-Cyd)} on solubility and percutaneous absorption of celecoxib through human skin.
6. Praveen Chaudhari et al. examined the cosolvency and cyclodextrins (CD) addition as a combined approach on the solubility of the hydrophobic drug, valdecoxib, attempted to increase the solubility of valdecoxib in water, using PEG-400, poloxamer-188 and 2 CDs (b-CD and Hp-b-CD).
7. Cinzia AnnaVentura et al. Prepared celecoxib-dimethyl-β-Cyclodextrin inclusion complex: characterized and studied the in vitro permeation. Where the ability of 2, 6-di-O-methyl-β-Cyclodextrin (DM-b-Cyd) to include the anti-inflammatory drug celecoxib (CCB) was evaluated.
8. Faiyaz Shakeel et al. studied the solubility and dissolution improvement of Aceclofenac using different nanocarriers. The aim was to improve solubility and dissolution of the lipophilic drug aceclofenac using three nanocarriers namely nanoemulsion, solid lipid nanosuspension and polymeric nanosuspension.
9. Swati Rawat et al. investigated the solubility enhancement of celecoxib using β-Cyclodextrin inclusion complexes. The formation of 1:1 complex with β-CD in solution was confirmed by phase solubility and spectral shift studies.
10. Neelam Seedher et al. examined the solubility enhancement of 4 cox-2 inhibitors, celecoxib, rofecoxib, meloxicam, and nimesulide, using a series of pure solvents and solvent mixtures.
DRUG PROFILE
* Name : Celecoxib
* Synonyms : Celebra, Celebrex
* Accession Number : DB00482
* CAS number : 169590-42-5
* Chemical Formula : C17H14F3N3O2S
* Structure : 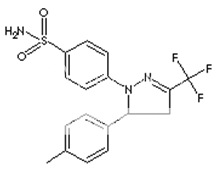
* Molecular weight : 318.372
* IUPAC Name : 4-[5-(4-methylphenyl)-3-(trifluoromethyl)- 1H-pyrazol-1-yl] benzene-1-sulfonamide
* Type : Non-steroidal Anti-inflammatory Drug (NSAID)
* Solvent solubility : Acetone, ethanol, ethyl acetate ,& Oils
* Appearance : White powder, Solid.
* Melting point : 157-158° C
* Dosage forms : Capsule
* Strength : Celebrex 50 mg, Celebrex 100 mg, Celebrex 200 mg, Celebrex 400 mg.
* Ionization constant (PKa): 11.1
* LogP : 3.99
* Storage : Store at room temperature between 15-30°c
* Absorption : Well absorbed in the gastrointestinal tract. When taken with a high fat meal, peak plasma levels are delayed for about 1 to 2 hours with an increase in total absorption (AUC) of 10% to 20%
* Bioavailability : Capsule—22-40% & solution—64-88%
* Protein binding : 97%, primarily to albumin and, to a lesser extent, œ1-acid glycoprotein
* Half life : Approximately 11hours
* Volume of distribution : 400 L
* Metabolism : Celecoxib metabolism mediated via cytochrome
* Route of elimination : Hepatic metabolism, unchanged drug recovered in the urine and faeces.
* Clearance : 500 ml/min
EXCIPIENT PROFILE [CYCLODEXTRIN]
Nonproprietary Names
BP: Betadex, PhEur: Betadexum, USPNF: Betadex
Synonyms
* Cyclodextrin : Cavitron; cyclic oligosaccharide; cycloamylose; cycloglucan; Encapsin; Schardinger dextrin.
* α-Cyclodextrin : Alfadex; alpha-cycloamylose; alpha-cyclodextrin;alpha- dextrin;CavamaxW6Pharma; cyclohexaamylose; cyclomaltohexose.
* β-Cyclodextrin : Beta-cycloamylose; beta-dextrin; Cavamax W7 Pharma; Cycloheptaamylose; cycloheptaglucan; cyclomaltoheptose; Kleptose.
* γ-Cyclodextrin : Cavamax W8 Pharma; cyclooctaamylose; gamma cyclodextrin.
Empirical Formula and Molecular Weight
* α-Cyclodextrin : C36H60O30 972
* β-Cyclodextrin : C42H70O35 1135
* γ-Cyclodextrin : C48H80O40 1297
Functional Category : Solubilizing agent; stabilizing agent.
Pharmaceutical Applications:
Cyclodextrins are crystalline, nonhygroscopic, cyclic oligosaccharides derived from starch. Among the most commonly used forms are a-, b-, and γ-Cyclodextrin, which have respectively 6, 7, and 8 glucose units. Cyclodextrins are ‘bucket like’ or ‘cone like’ toroid molecules, with a rigid structure and a central cavity, the size of which varies according to the cyclodextrin type. The internal surface of the cavity is hydrophobic and the outside of the torus is hydrophilic; this is due to the arrangement of hydroxyl groups within the molecule. This arrangement permits the cyclodextrin to accommodate a guest molecule within the cavity, forming an inclusion complex.
Cyclodextrins may be used to form inclusion complexes with a variety of drug molecules, resulting primarily in improvements to dissolution and bioavailability owing to enhanced solubility and improved chemical and physical stability. Cyclodextrin inclusion complexes have also been used to mask the unpleasant taste of active materials and to convert a liquid substance into a solid material. β-Cyclodextrinis the most commonly used cyclodextrin, although it is the least soluble. It is the least expensive cyclodextrin; is commercially available from a number of sources; and is able to form inclusion complexes with a number of molecules of pharmaceutical interest. However, β-Cyclodextrinis nephrotoxic and should not be used in parenteral formulations. β-Cyclodextrinis considered to be nontoxic when administered orally, and is primarily used in tablet and capsule formulations. β-Cyclodextrinderivatives tend to be nontoxic.
When used either orally or parenterally, and the derivatives 2- hydroxypropyl-β-Cyclodextrinand 3-hydroxypropyl-β-Cyclodextrinare becoming increasingly important in pharmaceutical formulations. α-Cyclodextrinis used mainly in parenteral formulations. However, as it has the smallest cavity of the cyclodextrins it can form inclusion complexes with only relatively few, small-sized molecules. In contrast, γ-Cyclodextrinhas the largest cavity and can be used to form inclusion complexes with large molecules; it has low toxicity and enhanced water solubility.
In oral tablet formulations, β-Cyclodextrinmay be used in both wet-granulation and direct-compression processes. The physical properties of β-Cyclodextrinvary depending on the manufacturer. However, β-Cyclodextrintends to possess poor flow properties and requires a lubricant, such as 0.1% w/w magnesium stearate, when it is directly compressed.
In parenteral formulations, cyclodextrins have been used to produce stable and soluble preparations of drugs that would otherwise have been formulated using a nonaqueous solvent. In eye drop formulations, cyclodextrins form water-soluble complexes with lipophilic drugs such as corticosteroids. They have been shown to increase the water solubility of the drug; to enhance drug absorption into the eye; to improve aqueous stability; and to reduce local irritation. Cyclodextrins have also been used in the formulation of solutions, suppositories and cosmetics.
Solubility:
α-Cyclodextrin: soluble 1 in 7 parts of water at 208C, 1 in 3 at 508C. β-Cyclodextrin: soluble 1 in 200 parts of propylene glycol, 1in 50 of water at 208C, 1 in 20 at 508C; practically insoluble
in acetone, ethanol (95%), and methylene chloride. γ-Cyclodextrin: soluble 1 in 4.4 parts of water at 208C, 1 in 2 at 458C.
Cyclodextrins should be stored in a tightly sealed container, in a cool, dry place.
Safety:
Cyclodextrins are starch derivatives and are mainly used in oral and parenteral pharmaceutical formulations. They are also used in topical and ophthalmic formulations.Cyclodextrins are also used in cosmetics and food products, and are generally regarded as essentially nontoxic and nonirritant materials. However, when administered parenterally, β-Cyclodextrinis not metabolized but accumulates in the kidneys as insoluble cholesterol complexes, resulting in severe nephrotoxicity. Other cyclodextrins, such as 2-hydroxypropyl-β-Cyclodextrin, have been the subject of extensive toxicological studies. They are not associated with nephrotoxicity and are reported to be safe for use in parenteral formulations.Cyclodextrin administered orally is metabolized by microflora in the colon, forming the metabolites maltodextrin, maltose, and glucose; which are themselves further metabolized before being finally excreted as carbon dioxide and water.
Cyclodextrins are not irritant to the skin and eyes, or upon inhalation. There is also no evidence to suggest that cyclodextrins are mutagenic or teratogenic.
Regulatory Status:
β-Cyclodextrinis included in the FDA Inactive Ingredients guide (IM, IV injections, and other injection preparations). Included in the Canadian List of Acceptable Non-medicinal Ingredients (stabilizing agent; solubilizing agent); and in oral and rectal pharmaceutical formulations licensed in Europe, Japan, and the USA.
AIM AND OBJECTIVES
The ability to deliver poorly soluble drugs will grow in the coming years as innovator companies rely upon NCEs for a larger share of the revenue within the pharmaceutical market. Similarly generic drug manufacturers will need to employ economically efficient delivery as more low solubility drugs go off patent, in order to maintain a competitive edge.
Poor solubility can hinder or even prevent drug development yet the volume and level of poorly soluble drugs is dramatically increasing leaving gaps in the development pipelines.
In the present study, Celecoxib taken (poorly soluble drug) as the model drug.
The main objective of the work to prepare inclusion complexes by various methodologies i.e. co-precipitation/co-evaporation, kneading, complexation etc. to enhance its aqueous solubility and in-vitro dissolution rate.
In present study betacyclodextrin has been selected as carrier because of their chemical and pharmacological inertness. Betacyclodextrin by virtue of their water solubility leads high degree of solubilization of poorly soluble drug.
PLAN OF WARK
I. Preparation of calibration curve by spectrophotometric method.
II. Preparation of physical forms.
III. Phase or saturation solubility studies.
IV. Drug content determination for the complexes.
V. Percentage yield determination
VI. In-vitro dissolution studies of complexes.
MATERIALS & EQUIPMENTS
MATERIALS:
Celecoxib was a generous gift from Cadila Health Care, Ankleshwar. β-cyclodextrin and HP β-cyclodextrin were purchased from Gangwal chemicals, Mumbai. All reagents were of analytical reagents grade. Distilled water was used for all the experiments.
LIST OF EQUIPMENTS USED:
|
Sr. No. |
Equipment Name |
Company [ Model ] name |
|
1 |
Sieve |
GEOSYN’S |
|
2 |
UV Visible Spectrophotometer |
ELICO SL159 |
|
3 |
Sonicator |
Pci |
|
4 |
Electronic Balance |
SAMSON |
|
5 |
Rota Shaker |
RS12REMI |
|
6 |
Hot Air Oven |
ROLEX |
|
7 |
Sigma Balance |
SIGMA 200/A |
|
8 |
Heating Mantle |
JSGW |
|
9 |
Dissolution Apparatus |
LABINDIA DISSO 2000 |
|
10 |
Mortar&Pestle |
---------- |
EXPERIMENTAL WORK
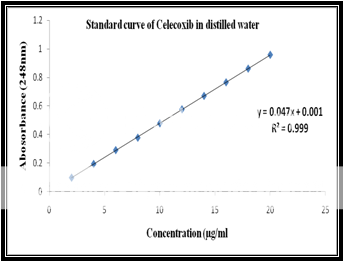
Calibration of celecoxib in distilled water at 248nm:
Standard stock solution:
Celecoxib (100 mg) was dissolved in 10 ml of methanol and then .the volume was made to 100 ml with distilled water to form a clear solution.
Working stock solution:
A series of Celecoxib solutions ranging from 2 to 20 mg/ml were prepared from standard stock solution. The low concentration was scanned in the range of 200-400 nm to get the maximum absorbance of 248 nm and the absorbances of the solutions were measured spectrophotometrically at 248 nm.
|
Celecoxib Concentration(ppm) |
Abs at 248nm |
|
2 |
0.098 |
|
4 |
0.194 |
|
6 |
0.291 |
|
8 |
0.378 |
|
10 |
0.479 |
|
12 |
0.577 |
|
14 |
0.671 |
|
16 |
0.766 |
|
18 |
0.862 |
|
20 |
0.959 |
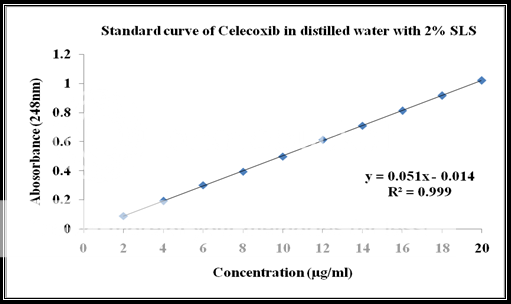
Calibration of celecoxib in distilled water at 248nm with 2% SLS:
Standard stock solution:
Celecoxib (100 mg) was dissolved in 10 ml of methanol and then .the volume was made to 100 ml with distilled water containing 2% SLS to form a clear solution.
Working stock solution:
A series of Celecoxib solutions ranging from 6 to 12 mg/ml were prepared from standard stock solution. The low concentration was scanned in the range of 200-400 nm to get the maximum absorbance of 248 nm and the absorbance of the solutions was measured spectrophotometrically at 248 nm.
|
Celecoxib Concentration(ppm) |
Absorbance at 248nm |
|
2 |
0.089 |
|
4 |
0.192 |
|
6 |
0.301 |
|
8 |
0.394 |
|
10 |
0.498 |
|
12 |
0.611 |
|
14 |
0.709 |
|
16 |
0.812 |
|
18 |
0.916 |
|
20 |
1.019 |
Preparation of physical forms:
The solid complexes of Celecoxib & β-CD/ HP β-CD (1:1 molar ratio) were Prepared by following methods:-
1. Physical mixture: The physical mixtures of Celecoxib & β-CD/HP β-CD [1:1 molar ratio] obtained by mixing pulverized powder (#100) together in pestle & mortar and designated as PM1 and PM2respectively.
2. Kneading method:
Celecoxib & β-CD/HP β-CD [1:1 molar ratio] was triturated in mortar and pestle and to it water was added and triturated to get a pasty mass. The wet mass was dried in oven at 450C and passed through sieve #100 and designated as KN1 and KN2respectively.
3. Coprecipitation method:
The aqueous solution of β-CD/ HP β-CD was added to an alcoholic solution of Celecoxib in 1:1 molar ratio. The resulting mixture was stirred for 1 hr & filtered and precipitate was dried at 45oC. The dried mass was pulverized & sieved through (#100), designated as CP1 and CP2 respectively.
All the formulations were kept in dessicator for 24 hour in order to remove any moisture present.
Evaluation of physical forms:
1. Saturation Solubility Studies
Excess amount of Celecoxib (pure drug) and the prepared physical forms were added to the glass vials containing 10 mL of distilled water. The sealed vials were shaken for 24 hr at 37oC followed by equilibrium for three days. Then, the aliquots were withdrawn through Whatman filter paper. The concentration of Celecoxib was determined by UV spectrophotometer at 248 nm.
2. Percentage yield
Percentage yield was calculated to know efficiency of methods,
Percentage yield = {Practical amount X 100}/Theoretical amount
3. Drug content
Celecoxib β-CD/HP β-CDcomplexes equivalent to 20mg of Celecoxib were weighed accurately and dissolved in 50 ml of methanol. Diluted suitably and drug content was analyzed at 248 nm by UV spectrophotometer.
4. In Vitro Dissolution studies
Invitrodissolution studies of the pure drug and the complexes with β CD & HP β CD were carried out with the following conditions:
USP Dissolution Apparatus : Type-II (Paddle Type)
RPM : 50
Dissolution medium : Distilled Water containing 2% SLS\
Volume of Dissolution Fluid : 900ml
Temperature : 37±0.5°C
Sample Size : 50 mg equivalent of Celecoxib
Sample of 5 ml was withdrawn at regular intervals and filtered by syringe filter of 0.45 µm. The withdrawn volume was replaced with fresh dissolution medium. The filtered samples were analyzed spectrophotometrically at 248 nm. Dissolution studies for each formulation were performed in triplicates.
Saturation Solubility in distilled water:
|
Formulations |
Solubility (mg/ml) |
|
Pure drug |
0.007 |
|
PM1 |
0.009 |
|
PM2 |
0.011 |
|
KN1 |
0.046 |
|
KN2 |
0.206 |
|
CP1 |
0.043 |
|
CP2 |
0.0795 |
Percentage yield and drug content from the physical forms:
|
Formulation |
% Yield |
% Drug Content |
|
PM1 |
89.93 |
81.62 |
|
PM2 |
87.59 |
84.36 |
|
KN1 |
82.61 |
78.72 |
|
KN2 |
81.03 |
80.52 |
|
CP1 |
76.42 |
|
|
PharmaTutor (ISSN: 2347 - 7881) Volume 1, Issue 1 Received On: 07/10/2013; Accepted On: 29/10/2013; Published On: 25/11/2013 How to cite this article: Panigrahi PP, Acharya AK, Solubility Enhancement Of Poorly Water Soluble Drug Celecoxib, PharmaTutor, 2013, 1(1), 53-64 |








