
{ DOWNLOAD AS PDF }
About Author:
Yogeshkumar B. Viradiya
Department of Regulatory Affairs, Institute of Management Sciences and Research (IMSR),
Nagpur, Maharashtra.
viradiya2210@gmail.com
Abstract:
Regulatory affair is the very important department in Pharmaceutical Company. For the protection of public health, government of various countries have developed the regulation for pharmaceutical, cosmetic product, pesticides, veterinary medicines, medical device, agrochemical and complementary medicines by controlling the safety and efficacy of product. Regulatory affair department prepare the registration document which submits to the regulatory agency of various countries for approval of new drug which contain the all important information about new drug. It is called the drug master file of Common technical document (CTD). Regulatory affair is the link between company and government authority. Regulatory affair is important for Product management, Clinical trial, Research & Development. Regulatory personnel have to maintain contact with other specialist like chemist, doctors, veterinarians, engineers, pharmacologist, toxicologist, pharmacists, and accountant. Regulatory authorities of different countries prepare their separate rules and regulation. Main aim of regulatory affair department is to provide safe and effective medicine to people of different companies. Separate rules and regulation in different countries which must be followed by all pharmaceutical company in all over the world.
Introduction:
Regulatory Affair: To protect public health, government of various countries have developed the regulation for pharmaceutical, cosmetic product, pesticides, veterinary medicines, medical device, agrochemical and complementary medicines by controlling the safety and efficacy of product.1
The companies make a worthwhile contribution to public health and welfare, by supplying the product that are safe and are of standard quality. To fulfil these requirements the companies undergo discovery, testing, manufacturing and marketing of these product. Most companies either small medium or multinational pharmaceutical corporation or innovative biotechnology companies have special department of Regulatory Affair. If the company doesn’t have the regulatory affair department then they have to rely on expert advice or regulatory consultant, but they must comply all the requirements essential for drug regulation.1
Main concern of Regulatory affairs Professional’s is: 1
· To keep record of all drugs
· To keep registration documents.
· Obtaining and maintaining marketing authorisation of product
· Presenting these registration documents to regulatory for the product
· To tract the ever-changing legislation in the entire region, in which the company wishes to distribute its products.
· Advice the higher authority on legal and scientific requirements
· Collect and evaluate the scientific data that their research and development colleagues are generating
· They play important contribution both commercially and scientifically starting from the beginning of the development of a product till the marketing of product.1
Importance of Regulatory Affair (RA)1
· The Regulatory Affairs department is the first point of contact between the government authority and the company.
· Play an important part in coordination scientific endeavour with regulatory demands.
· Maximize cost effectiveness of company’s resource
· Communicate the government official perception to the company
· Influence the strategic decision of their companies.
· Help to launch the drug in market on time, because a small delay can affect the financial status of company.
· Report the accurate marketing data to the higher authority.1
Link of regulatory affair department with other department:1
Regulatory department is the centre of any organisation and regulatory specialists have to work with co ordination of other department specialist. Regulatory personnel have to maintain contact with other specialist like chemist, doctors, veterinarians, engineers, pharmacologist, toxicologist, pharmacists, and accountant.
The regulatory department work as a bridge in following way:1
1. The Regulatory specialist collects the information from all other department and presents that information to regulatory authority.
2. This gives an idea of works of other either they are functioning properly or maintaining the standard or they have to make improvement.
3. Pass the feedback of regulatory department to the rest of the company
4. Maintaining advertising and marketing regulations
5. Evaluate the staff and assign the work according to requirements of regulatory bodies1
International Regulatory Environment2
Good Manufacturing Practices has been in practice from Old Testament times (Laws of Kashrut). The Nuremberg Code, 1947 on Permissible Medical Experiments provided for basic principles to conduct medical experiments on human beings followed by Declaration of Helsinki (1964), Belmont Report of
USA6 (1978) and WHO GCP (1995) and ICH GCP in 1996. In 1959, Canada instituted its QUAD regulations, which is the first recognizable drug GMP of modern era. It was followed by GMPs of USA in 1963 and that of UK in 1972.
Today 35 member countries along with 11 candidate countries and 4 international agencies have joined together to create the Pharmaceutical Inspection Cooperation Scheme (PIC/S) to promote a globally accepted GMP. The International Conference on Harmonization (ICH) was established in 1990
and has succeeded in harmonizing GMPs for manufacture of Active Pharmaceutical Ingredient (API), validation of analytical methodology, guidelines for performance of stability studies, harmonization of pharmacopoeal monographs and test methods and other guidelines of working of GMP.2
Regulatory Affairs in Product Management2
The key role of RA professional is broader than registration of products, they advise companies both strategically and technically at the highest level. Their role begins right from development of a product to making, marketing and post marketing strategies. Their advice at all stages both in terms of legal and
technical requirements help companies save a lot of time and money in developing the product and marketing the same. For countries that do not have their own regulations the World Health Organization guidelines on health matters and World Trade Organization on trade regulations between nations is followed.2 Product management team which decide the strategy after concerning with the regulatory affair department.
Regulatory Affairs in Clinical Trials2
The RA professional is the primary link between the company and worldwide regulatory agencies such as US Food and Drug Administration (USFDA & Centre for Devices and Radiological Health), Medicines and Healthcare Products Regulatory Agency, United Kingdom, (UKMCA), Therapeutic
Goods Administration, Australia, European Medicines Agency, Organization of Economic Collaboration and Development (OECD) and Health Canada. He also communicates and interprets the seemingly endless mace of laws, regulations and guidelines to the other departments of the company. The RA personnel develops strategies to overcome delays and presents finding of clinical trials to the regulatory bodies so as to get quick clearance thus reducing the time for approval of new molecules. At its core, the RA professional facilitates the collection, analysis and communication about the risks and benefits of health products to the regulatory agencies, medical and health systems and the public. Operationally RA is responsible for assuring that government obligation, market driven demands and evolving scientific conventions are understood and addressed by various stakeholders.2
Regulatory Affairs in R&D2
The regulatory affairs personnel work hand in hand with marketing and R&D to develop, innovative products that take advantage of new technological and regulatory developments to accelerate time to market. With new products expected to add significant revenues to the company’s bottom lines, small decreases in time to market equate to large material gains in revenue and profit. Employing adaptive clinical trial strategies, obtaining quick approval from regulatory authorities and avoiding pitfalls in processes can accelerate development of new products and help to reduce costly errors and time lags.2
Challenges to regulatory affair Professionals:3
· Multidimensional
· Knowledge in science and technology
· Prolific communication skill
· Deal with people with different background, skill, culture and personalities
· Deal with conflicting loyalties, Motivation, social and ethical responsibilities.3
Total Regulatory:
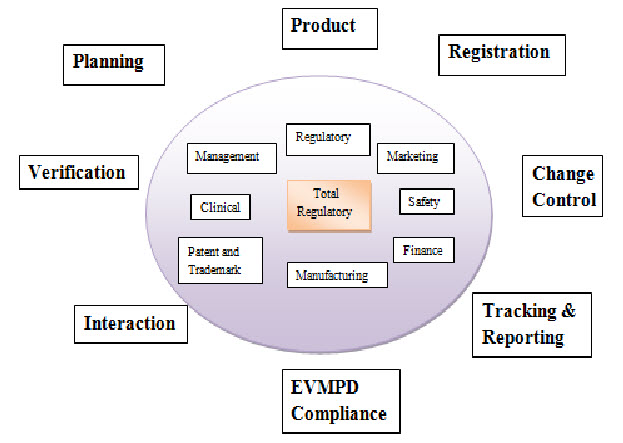
Figure: 1: Total Regulatory4
Total Regulatory Affair in Which: Regulatory, Marketing, Safety, Finance, Manufacturing, Patent and Trademark, Clinical, Change control, Tracking and Reporting, EVMPD Compliance, Interaction, Verification, Planning, Product.
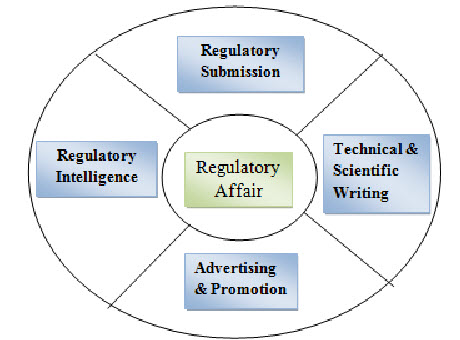
Figure: 2 Regulatory Affairs5
The main function of Regulatory Affairs:
Preparation of drug master file, dossier, Common technical document CTD, Electronic common technical document eCTD.
eCTD: eCTD is the electronic form of document which directaly send to the regulatory authority of different countries on their email address or send in to the DVD which is very easy and saving of paper.
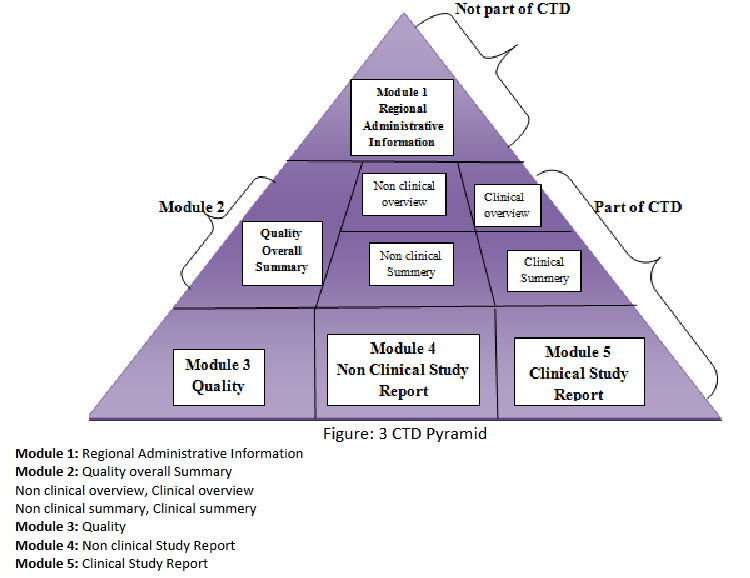
Figure: 3 CTD Pyramid
Module 1: Regional Administrative Information
Module 2: Quality overall Summary
2.1 Non clinical overview, Clinical overview
2.2 Non clinical summary, Clinical summery
Module 3: Quality
Module 4: Non clinical Study Report
Module 5: Clinical Study Report
Drug Master File: Drug Master File (DMF) is a submission to the Food and Drug Administration (FDA) that may be used to provide confidential detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging and storing of one or more human drugs. The submission of a DMF is not required by law or FDA regulation. A DMF is submitted solely at the discretion of the holder. The information contained in the DMF may be used to support an Investigational New Drug Application (IND), a New Drug Application (NDA), an Abbreviated New Drug Application (ANDA), another DMF, an Export Application, or amendments and supplements to any of these. A DMF is NOT a substitute for an IND, NDA, ANDA, or Export Application. It is not approved or disapproved.6
Technical contents of a DMF are reviewed only in connection with the review of an IND, NDA, ANDA, or an Export Application.
This guideline does not impose mandatory requirements (21 CFR 10.90(b)). It does, however, offer guidance on acceptable approaches to meeting regulatory requirements. Different approaches may be followed, but the applicant is encouraged to discuss significant variations in advance with FDA reviewers to preclude spending time and effort in preparing a submission that FDA may later determine to be unacceptable. Drug Master Files are provided for in 21 CFR 314.420. This guideline is intended to provide DMF holders with procedures acceptable to the agency for preparing and submitting a DMF. The guideline discusses types of DMF's, the information needed in each type, the format of submissions to a DMF, the administrative procedures governing review of DMF's, and the obligations of the DMF holder.6
DMF's are generally created to allow a party other than the holder of the DMF to reference material without disclosing to that party the contents of the file. When an applicant references its own material, the applicant should reference the information contained in its own IND, NDA, or ANDA directly rather than establishing a new DMF.6
TYPES OF DMFs6
Type I: Manufacturing Site, Facilities, Operating Procedures, and Personnel Type II: Drug Substance, Drug Substance Intermediate, and Material Used in Their Preparation, or Drug Product
Type III:Packaging Material
Type IV:Excipient, Colorant, Flavor, Essence, or Material Used in Their Preparation
Type V:FDA Accepted Reference Information
Regulatory Environment in Different Countries:
Regulatory environment in different countries of world is different according to their rules and regulation of regulatory bodies.
Regulatory environment in India7
Central drug standard control organisation (CDSCO) and the state licensing authority issue the licenses for import, manufacture, sale and testing of drug and cosmetics. The issuing of drug manufacturing licences is a vital function of a regulatory body of any country because the determines the quality and availability of drugs in market.The Drug and Cosmetic Act 1940 and Rules 1945 were passed by the India's parliament to regulate the import, manufacture, distribution and sale of drugs and cosmetics. The Central Drugs Standard Control Organization (CDSCO) and the office of its leader, the Drugs Controller General (India) [DCGI] were established. In 1988, the Indian government added Schedule Y to the Drug and Cosmetics Rules 1945. Schedule Y provides the guidelines and requirements for clinical trials, which was revised in 2005 to bring it is internationally accepted process. The changes includes, establishing definitions for Phase I-IV trials and clear responsibilities for investigators and sponsors. In 2006 clinical trial was divided into two categories. (category A) clinical trials can be conducted in another markets with competent and mature regulatory systems whereas the remaining ones fall in to another category (category B) Other than A. Clinical trials of category A (approved in the U.S., U.K, Switzerland, Australia, Canada, Germany, South Africa, Japan and European Union) are eligible for fast tracking in India, and are likely to be approved in eight weeks. Category B of the Clinical trials is under more scrutiny, and approve within 16 to 18 weeks.The confirmatory trials (Phase III) are conducted to generate data regarding the efficacy and safety of the drug in 100 patients (in 3-4 canters) to confirm efficacy and safety claims. Phase III trials should be conducted on a minimum of 500 patients spread across 10-15 centres, If the new drug substance is not marketed in any other country. The new drug registration (using form # 44 along with full pre-clinical and clinical testing information) is applied after the completion of clinical trials. The comprehensive information on the marketing status of the drug in other countries is also required other than the information on safety and efficacy. The information regarding the prescription, samples and testing protocols, product monograph, labels, and cartons must also be submitted. The application can be reviewed in a range of about 12-18 months. An application to conduct clinical trials in India should be submitted along with the data of chemistry, manufacturing, control and animal studies to DCGI. The date regarding the trial protocol, investigator's brochures, and informed consent documents should also be attached. A copy of the application must be submitted to the ethical committee and the clinical trials are conducted only after approval of DCGI and ethical committee.7
Regulatory environment in USA8
In the US, drugs are regulated by the Food and Drug Administration (FDA [fda.gov/]). The US drug approval process is considered to be one of the most stringent in the world. The reviewers are very thorough and often reanalyze the data to ensure that it supports the same conclusions reached by a sponsor.
The Federal Food, Drug and Cosmetic Act states that a new drug may not be introduced into interstate commerce unless the FDA has approved a New Drug Application (NDA) for it (21 U.S.C. 355). Every new drug must receive marketing approval from the FDA prior to commercialization. The NDA consists of all the information and data that have been collected on a drug during its development. The goals of the NDA are to permit the FDA reviewer to reach conclusions on the following three key areas:
1. Whether the drug is safe and effective in its proposed use, and whether the benefits outweigh the risks.
2. Whether the drug’s proposed labelling (package insert) is appropriate, and what it should contain.
3. Whether the methods used in manufacturing the drug and the controls used to maintain the drug’s quality are adequate to preserve the drug’s identity, strength, quality and purity.1
The documentation that is submitted in the NDA should explain the whole process of drug development including all animal and human studies that have been performed; how the studies were designed and analyzed and the results thereof; the ingredients of the drug; how the drug is manufactured, processed and packaged; and its stability.
It is important to consider the views of the FDA and the goals of an NDA very early in the development of a new drug. For instance, clinical trials must support the claims made with regard to safety and effectiveness. As well, the proposed labelling must be supported by the data from the clinical trials. New claims that are not fully supported by the data package are not likely to be approved. As equally important as the clinical data are the chemistry, manufacturing and controls. Regardless of its safety and efficacy, a new drug may not receive approval if the manufacturing and controls processes cannot ensure consistent quality across numerous batches.8
Regulatory environment in European Union
Regulatory Agency Of European Union
European medicine agency (EMA)
European directorate for the quality of medicine (EDQM)
Types of process
Nationalized process
Decentralized process
Mutual recognition process
Centralize process
In European Union (EU), all European countries which are come in the European Union. There are no specific rules & regulation for different countries, same rules & regulation follow by all European countries for pharmaceutical. New drug registration process is same for all countries. All medical products were approved for marketing at the National level. The first reorganization procedure was introduced in 1983 and a single national review in case of pharmaceutical/medicinal product for marketing authorizations in all EU's countries was made feasible. The first aim of this procedure was to create a united standard for product review among national regulatory authorities.
A sponsor has several options when seeking approval to market a new drug in Europe: a national authorization procedure, a decentralized procedure, a mutual recognition procedure, or a centralized procedure. Products that must use the centralized procedure include the following:9
• All biologic agents or other products made using high-technology procedures
• Products for HIV/AIDS, cancer, diabetes, neurodegenerative diseases, auto-immune and other immune dysfunctions and viral diseases
• Products for orphan conditions9
Regulatory environment in South East Asia10
South East Asia is a growing pharmaceutical market. The regulatory environment has similar characteristics but drug registration requirements and processes differ among the countries. An ASEAN initiative to harmonize the requirements for drug registration is in progress. Many multi-national research-based pharmaceutical companies have begun to conduct multi-center trials in their global drug development programs involving Asian medical centers for faster patient recruitment to achieve faster drug development. In the recent years, there is a steady increase in clinical trial activities in South East Asia.9
|
Countries |
Drug Regulatory Authorities |
|
Indonesia |
National Agency of Drug & Food Control |
|
Malaysia |
Drug Control Authority, NCE Unit |
|
Thailand |
ThaiFDA, Drug Control Div |
|
Philippines |
BFAD, DoHT |
|
Singapore |
Health Sciences Authority (HSA) |
|
|
Centre for Pharmaceutical Admin, (CPA)Centre for Drug Evaluation (CDE) |
Regulatory environment in ROW countries11
Product registration in rest of world is a challenging task like regulated countries (US, EU & Japan) as they are not harmonized. It creates a difference in regulatory environment in Semi Regulated countries. Enormous diversity of regulatory requirements is found in this area. This region consists of mainly the countries from Asia pacific, Latin America, Eastern Europe, Africa and Gulf countries. Countries from Asia pacific and Gulf have somewhat harmonized their regulatory environment through The Association of Southeast Asian Nations (ASEAN) and Gulf Co-operation Council (GCC) organizations, ROW countries yet to be harmonized regulations in their respective regions. The urgent requirement to rationalize & harmonize regulation was required by instance of rising cost of health care, research & development and to meet the public requirement for safe and efficacious treatments to patient in need. ICH committee has given priority to harmonize the format of reporting data for quality, safety and Efficacy in the application dossier. The commercial significance of ROW markets is increasing globally. It is crucial that pharmaceutical companies keep up-to-date with the latest regulatory developments to ensure their place on the ROW market.11
Regulatory environment in Australia12
|
Manufacturers |
To manufacture the medicine in Australia or overseas |
|
Sponsors |
To import, export, or manufacture Medicines. An Australian manufacturer may also act as a sponsor. |
|
Therapeutic Goods Administration (TGA) |
Government body that has regulatory control of therapeutic goods (including medicines) in Australia |
|
Agents Consultants |
such as PharmOut, that act on behalf of manufacturers or sponsors, to register medicines in Australia |
In order to register a medicine in Australia you need to ensure that the manufacturer is licensed and in compliance with the requirements of Good Manufacturing Practice (GMP).
You also need to:
* assess the risk of your medicine
A: - high-risk medicines must be assessed for safety, efficacy and quality and registered on the Australian Register of Therapeutic Goods (ARTG)2 before supply in Australia
B: - low-risk medicines must be assessed for safety and quality and listed on the ARTG before supply in Australia
* implement post market surveillance systems and adverse event reporting Programs
* ensure that advertising and labelling is performed in accordance with the
* Therapeutic Goods Advertising Code.12
Parts which Cover under the Regulatory Affairs:
· Good Manufacturing Practices
· Good Laboratory Practices
· Good Clinical Practices
· Validation
· Drug Master File
· Registration application for approval of IND,NDA,ANDA
· Quality Assurance
· Quality Control
International Regulatory Market Divided in Various Types:11
Regulated Market: US, EU (UK, France, Ireland, Sweden, Germany etc.), Japan, Canada, Australia, New Zealand, South Africa
Semi-Regulated Market:
Asian Countries:
(a)Asia (India, Bangladesh, Sri Lanka)
ASEAN: 10 Countries group - Philippines, Vietnam Singapore, Malaysia, Thailand, Indonesia, Laos, Cambodia, Brunei Darussalam, Myanmar
(b)African countries (Algeria, Zambia, Ethiopia, Ghana, Kenya, Malawi, Mozambique, Namibia, Nigeria, Sierra Leone, Tanzania, Zimbabwe etc)
(c)Middle East countries (Gulf Co-operation Council countries i.e. Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, UAE)
(d)Latin America (Mexico, Brazil, Panama, Peru, Guatemala, Argentina, Chile, Dominican Republic)
(e)CIS (common wealth of independent states): Russia, Ukraine, OFSUs (Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kirghizstan, Moldova, Tajikistan, Turkmenistan, Uzbekistan etc.)
Why is Regulatory Affairs Needed?13
· Design = Development Plan
· Co-ordination= Writing/reviewing, supervising
· Construction= Assembling & Submission Management
· Testing= Where are the weaknesses?
Drug regulations:
· National Laws (e.g. India- CDSCO)
· National and Regional Guidelines
· Regional Laws
· International Guidelines (ICH, WHO)
A Strong Career in Regulatory Affair
· 1-4 years executive level
· 4+ years senior level
· Associate director
· Director
· Vice President
· President
Training in Regulatory Affair
· On the job training
· Regulatory Affairs Specialised Course
· Internal Company training
· External Company training & Workshop
Opportunities13
· International Opportunities
· Projects, secondments, relocation
· Shaping the future within companies
· Outside industry
· Trade bodies
· Become a regulator!
· Sub-specialties
· Regulatory Operations
· Regulatory Intelligence
· Other industry disciplines
Need for good regulatory affair professional?1
1. Must be graduate in science either pharmacy or biotechnology or life science
2. Good communication skill to present oral and written evidence in front of expert team of higher authority of company doctors, pharmacists, scientist.
3. An analytical mind.
4. Technical skill to tackle today’s ever changing policies
5. Ability to evaluate the strength and weakness of the technical and legal option on which the success of company depends
6. Management skill that help to achieve the challenging gaols
7. Ability to quickly new concept
8. Can work as part of multidisciplinary team and lead them when necessary
9. Can assure the authority regarding safety, efficacy and quality of the product.
Academic knowledge
Post Graduate in Pharmacy with Pharmaceutical Administration and Management or Regulatory Affairs specialization will be the preferred qualification to qualify for as a RA professional. In the curriculum should have covered topics such as Handling laboratory and manufacturing deviations, Pre approval inspections, Total Quality management, GMP Certification and enforcement actions, Maintenance and Update of Product Master Files, Internal Compliance of Documentation, , Method Validations, Process Validations, Master Validation Plan, Protocols, Standard Operating Procedures (SOPs), Auditing and Compliance Functions, Regulatory strategies, Regulatory agencies, legislation and documentation systems, Coordination and Assembling of Common Technical Document (CTD/eCTD), Quality systems, Quality Assurance as required for USFDA, UKMCA/UKMHRA, MCC, WHO etc., FDA/UKMHRA queries and submission, application requirements and guidelines, electronic submissions, medical device regulations, stability as per ICH guidelines & Multi-brand registrations (MBRs);, Toxicological and Clinical Trial Information, Re-registration Documents Design, Role of the International Business Operations of the Pharmaceutical MNCs in Attracting the FDI, Clinical Pharmacy, Drug Trials and Vaccine Trials Guidelines, Drug Laws , Investigational New Drug Applications, International harmonization, practice of regulatory affairs, USP Pharmacological , Formatting, assembling and submitting the New Drug Applications, Human Genetic Research, Clinical Trials, Indian Ethics Committee, Good Clinical Practices (GCP), Pharmaco-vigilance and Adverse Drug Reactions reporting, Clinical Trial Regulation, Intellectual Property Rights, Basis of Patentability, Patent Application Procedure, Compulsory License, Infringement of Patents, Product Registration for Regulated and Non Regulated Markets etc.
How to join Regulatory Affair:3
· Regulatory affair job is boring but good profession.
· Regulatory education and work experience would help to be a successful Regulatory Affairs profession.
· For entry in Regulatory profession small company would get the better knowledge about regulatory affair.
List of Countries and their Regulatory Authority
|
Country |
Name of Regulatory Authority |
|
USA |
Food and Drug Administration (FDA) |
|
UK |
Medicines and Healthcare Products Regulatory Agency (MHRA) |
|
India |
Central Drug Standard Control Organization (CDSCO) |
|
Australia |
Therapeutic Goods Administration (TGA) |
|
Europe |
European Medicines Agency (EMEA) |
|
Canada |
Health Canada |
|
Costa Rica |
Ministry of Health |
|
New Zealand |
Medsafe - Medicines and Medical Devices Safety Authority |
|
Netherlands |
Medicines Evaluation Board |
|
Denmark |
Danish Medicines Agency |
|
Sweden |
Medical Products Agency (MPA) |
|
Ireland |
Irish Medicines Board |
|
Italy |
Italian Pharmaceutical Agency |
|
Ukraine |
Ministry of Health |
|
Nigeria |
National Agency for Food and Drug Administration and Control (NAFDAC) |
|
Singapore |
Centre for Pharmaceutical Administration Health Sciences Authority |
|
Hong Kong |
Department of Health: Pharmaceutical Services |
|
Sweden |
Medical Products Agency (MPA) |
|
Paraguay |
Ministry of Health |
|
China |
State Food and Drug Administration |
|
Thailand |
Ministry of Public Health |
|
Germany |
Federal Institute for Drugs and Medical Devices |
|
Malaysia |
National Pharmaceutical Control Bureau,Ministry of Health |
|
Pakistan |
Drugs Control Organization, Ministry of Health |
|
South Africa |
Medicines Control Council |
|
Sri Lanka |
SPC,Ministry of Health |
|
Switzerland |
Swissmedic , Swiss Agency for Therapeutic Products |
|
Japan |
Ministry of Health, Labour & Welfare(MHLW) |
|
Brazil |
Agencia Nacional de Vigiloncia Sanitaria (ANVISA ) |
|
Uganda |
Uganda National Council for Science and Technology (UNCST) |
INTERNATIONAL ORGANIZATIONS
World Health Organization (WHO)
World Trade Organization (WTO)
International Conference on Harmonization (ICH)
World Intellectual Property Organization (WIPO)
Pan American Health Organization (PAHO)
Special line about Regulatory Affairs:13
· Every product is different
· Pharmaceutical, Non-clinical and Clinical issues emerge in all stages of development
· Regulatory guidelines don’t always address problems encountered
· Discussion with the Regulatory Agencies is usually required at some point
Conclusion
This study shows that regulatory affair is very important for all pharmaceutical companies around the world. The main aim of regulatory affair department is to give safe and effective medicine to the people around the world. Regulatory Affair is also a good profession for students who are engaged in sturdy of Bachelor of Pharmacy or other science field. In this study we show the duties and responsibility of regulatory affair professional. Drug regulatory agency of various countries which give the strict rules and regulation which must be followed by pharmaceutical companies. A regulatory affair is also important for the Research and development, Product management, Clinical trial, marketing authorisation. Becoming a good regulatory affair officer executive some special skill which needed like sound knowledge about regulatory affair, good communication skill, knowledge about drug laws. All pharmaceutical companies have their own regulatory affair department those who don’t have they concerts with regulatory consultancy for product approval and marketing authorisation.Post Graduate in Pharmacy with Pharmaceutical Administration and Management or Regulatory Affairs specialization will be the preferred qualification to qualify for as a RA professional.
References:
1. A Regulatory affair book published by IMSR Nagpur.
2. hygeiajournal.com/downloads/554336197subash%20,ansa.pdf
3. Dr.Murlidhara_Gavini.pdf, vpmthane.org/adc/DRA_Seminar_04.12.2010/Dr.Muralidhara_Gavini.pdf
4. arisglobal.com/regulatory-affairs/
5. viglya.com/regulatory-affairs/
6. Guidance(Drugs)_Drug Master File_Guidelines.pdf
URL: fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122886.htm
7. NEW DRUG APPROVAL PROCEDURE IN INDIA_Pharmatutor.pdf URL: http://www.pharmatutor.org/articles/new-drug-approval-procedure-india?page=0,2
8. How_New_Drug_Approved_in_US.pdf URL: marsdd.com/dms/entrepreneurtoolkit/Regulatory-PDFs/How_New_Drugs_Approved_in_US.pdf
9. How_New_Drug_Are_Approved_In_Europe.pdf URL: marsdd.com/dms/entrepreneurtoolkit/Regulatory-PDFs/How_New_Drugs_Are_Approved_in_Europe.pdf
10. Regulatory Environment and Clinical Trial in South East Asia.pdf URL: cde.org.tw/Data/CDEDoc/Documents/Regulatory%20Enviroment%20and%20Clinical%20Trials%20in%20South%20East%20Asia.pdf
11. EXPORT REGISTRATION OF PHARMACEUTICALS IN REST OF WORLD COUNTRIES (ROW), URL: 391-1253-1-PB.pdf
12. White_paper_how_to_register_medicines_australia.pdf URL: pharmout.net/downloads/white_paper_how_to_register_medicines_australia.pdf
13. networkpharma.com/slides/meldrum_020608.pdf
REFERENCE ID: PHARMATUTOR-ART-2174
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 6 Received On: 02/04/2014; Accepted On: 06/04/2014; Published On: 01/06/2014 How to cite this article: Y Viradiya; Regulatory Affair: Link between company and Government Authority; PharmaTutor; 2014; 2(6); 9-20 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








