
About Authors:
Madhulika Kabra1, Pramod Khatri2*, Neerja Gupta3
1 Professor of Pediatrics,
2,3 Department of Pediatrics,
AIIMS Hospital, India
*pramodsinghkhatri@gmail.com
Abstracts:-
As experts strive for more superior effectiveness throughout the drug development process, it is crucial that they adventure all accessible qualified information to guide them. It is even more common to see drug discovery program disclosure support utilization of structural biology to improve the quality of molecular design.
REFERENCE ID: PHARMATUTOR-ART-1820
Introduction:-
Structure-based drug -design (SBDD) makes the most of the capability to confirm and visualize the 3D structure of drug target molecules. The crystal structure of a lead molecule bound to the objective of target might be important in controlling the outline of adjustments to enhance the compound's affinity for the target, or its selectivity versus different targets. Such structural informative data has been utilized successfully in design of several approved medications. SBDD could be utilized throughout the drug discovery process and is of specific utility throughout hit identification and the lead-optimization process.
Hit identification
Identification of excellent-hits might be a challenge for any system, yet in particular for novel target classes for example those being researched in the emerging territory of epigenetics. Structural learning of the target permits the utilization of virtual screening to distinguish hits for proteins that have no known inhibitors, or to gain access to novel binding sites on a known target. Virtual screening compares the 3D compliance of a potential inhibitor with the target binding site, to evaluate the level of spatial and electrostatic complementarity.
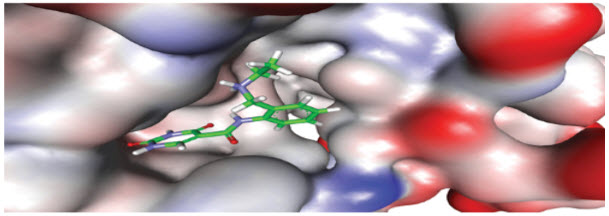
Figure 1. Virtual screening. Cropping of a small-molecule into the binding site of the target. (Source: Domainex)
Just those compounds that are anticipated to bind well—known as -"virtual hits"—need to be advanced into biological screens. The quality of applying structural learning is the elimination of large number of unacceptable compounds, consequently enhancing the screening library and improving the hit-rate1. While hits may be produced by random screening of compound libraries, this methodology is less proficient. e.g., to find hits for the lysine methyltransferase targets G9a and SMYD2, two groups of analysts have reported screening countless compounds , however yielded a couple of validated hits.2,3
Lead optimization
Once a hit progression has been distinguished, structural informative data empowers the coupling mode of the inhibitor or ligand to be verified, the rationalization of structure-activity relationships (SAR), and the direction of outline effort towards a more optimal connection with the active site. Samples of SBDD from the recent literary works uncovering clinical candidates incorporate recognizing key connections and bolting the bioactive conformation to gain strength through recognizing drug-like bio-isosteres, as in linsitinib/OSI-906, an oral inhibitor of IGF-1R and IR as of now in a Phase 3 clinical trial in adrenocortical carcinoma;4 steering drug design into unexploited areas of the focus as in PF-489791, a PDE5 inhibitor presently in Phase 2 for Raynaud's disease;5 and, streamlining the framework while holding key interaction, as in Xalkori/crizotinib, approved in 2011 to treat certain patients with late-stage lung cancer.6
Structural qualified information is also used to enhance selectivity over identified targets , an area which has recently been completely reviewed.7 Structural qualified data has come to be so precious to drug design that if a crystal of the target is not ready, a homology model will frequently be manufactured and utilized as a surrogate, for example for EZH2, an epigenetic target of great interest.8
Experiments in protein expression
In a perfect world, one might like a crystal structure of the focus at the start of a project, preferably holding a bound ligand, yet consistently this informative data just arrives later in the discovery program. A normal explanation behind this bottleneck in creating high-resolution crystal structures is the need for hefty amounts of pure, dissolvable protein that will promptly shape crystals. Full-length proteins are regularly too extensive and complex to process in recombinant cell expression frameworks and could be challenging—or even difficult—to solidify. An elective to utilizing the full-length protein is to focus just on the area of specific concern, for example the significant synergist or tying domain. The test then comes to be the means by which to recognize a piece of the protein that is simpler to control and express, yet holds the fitting amino-acid sequence that permits the protein to be folded rightly, and in certain cases, to have functional movement.
The most extensively utilized strategy to distinguish these areas is bioinformatics investigation of the gene and the protein it encodes.9 In silico examination and expectation of suitable expression builds might be supplemented by test strategies for example fractional proteolysis.10 However, domain limits recognized through bioinformatics are not vitally optimal for expression; a couple of amino acid deposits can altogether change the dissolvability, stability, and the level of expression of the hunted protein. Bioinformatics methodologies can't anticipate domain limits with sufficient accuracy to give constantly dependable outcomes. Consequently the traditional methodology is an iterative, trial-and-slip procedure of planning and redesigning builds until a sufficiently exceptional build is discovered, or the hunt must be surrendered.
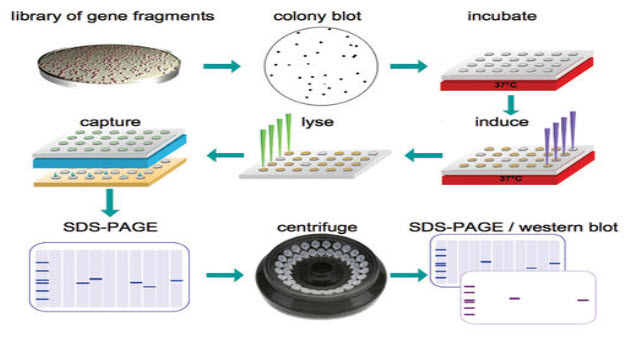
Figure 2. The Combinatorial Field Hunting screening process. A reference library of 20,000 to 100,000 E. coli clones, each holding a DNA fragmentation from the target gene is subjected to colony hybridization utilizing an immune response that recognizes poly-histidine tags appended to the expressed protein feature. Colonies expressing his-tagged dissolvable protein are chosen and expressed on a large scale. Solvent extracts are analyzed and clones that generate great levels of recombinant protein are subjected to centrifugation to uproot aggregated protein. Clones are end-sequenced to affirm their character. Protein fragments equipped to tie to an known ligand might be utilized to develop binding or activity assays.
An elective approach is to quickly produce a hefty number of randomly divided construct from the gene of interest, and to express and select those develops that encode exceptionally expressed, stably-folded dissolvable proteins.11,12 When properly executed, this impartial approach of random fragmentation and screening is fast, gives an clear cut results, and has the point of interest that it can recognize expressible locales inside a target gene that are surprising or unpredicted. It is additionally suitable for handling proteins with unfamiliar or crudely comprehended domain architecture. Library-based methodologies test large amounts of fragments, order of extent more terrific than the conventional iterative methodology. Hence, the articulation space might be inspected exhaustively and such fine-testing may be specifically convenient for focuses on whose expression is delicate to the exact sequence at the N-and C-termini.
The Combinatorial Domain Hunting approach to protein interpretation
In a recent case analysis, Brussels-based pharmaceutical association UCB and Domainex utilized an randomized fragmentation procedure reputed called to be Combinatorial Domain Hunting (CDH) to distinguish a protein develop suitable for structure-based drug design, for a target that had previously proved difficult to express and crystallize.13 Figure 2 exhibits how once a library of DNA fragments has been produced utilizing the CDH random fragmentation approach, the clones that code for stably-folded, dissolvable domain are recognized and are chosen for the next phase of medication discovery.14
The protein, mitogen-activated protein kinase (MAPKK, otherwise called MEK) is of major therapeutic interest. Even though one crystal structure of this kinase had been accounted for at the beginning of the project, endeavors to enhance the expression yield were unassuming, and efforts to generate structures of MAPKK-inhibitor edifices were unsuccessful. The CDH random fragmentation approach recognized a construct that cover the kinase domain and takes shape essentially superior to the bioinformatically designed expression constructs. The develop recognized by CDH as giving the most astounding outflow levels of solvent protein held an unexpected N-terminal expansion notwithstanding the center domain. The protein dominion distinguished utilizing the CDH methodology was accordingly used to resolve the structure of MAPKK and various MAPKK-inhibitor co-structures. The structural information data supported UCB researchers in outlining a novel class of MEK-1 inhibitors.15
Protein-protein collaborations are another especially testing region for medication discovery, and various scientists accept that these targets will frequently just be tractable through access to structural informative content. To that end a variant of the CDH engineering, called CDH2, which permits the recognizable proof of heterodimeric domain buildings, has been developed.16 These advances will permit drug scientists utilizing SBDD to stay at the cutting edge of experimental discovery.
Competing interests:-
The authors declare that they have no competing interests
References:-
1. Jenkins JL, et al. Proteins: Structure, Function, and Bioinformatics, 2003;50(1),81–93.
2. Kubicek S, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007; 25:(3),473–481.
3. Ferguson AD, et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 2011;19(9):1262-73.
4. Mulvihill and Buck. The discovery of OSI-906, a small-molecule inhibitor of the insulin-like growth factor-1 and insulin receptors. Accounts in Drug Discovery: Case Studies in Medicinal Chemistry. 2011; 71-102.
5. Bell and Palmer. The discovery of the long-acting PDE5 inhibitor PF-489791 for the treatment of pulmonary hypertension. Accounts in Drug Discovery: Case Studies in Medicinal Chemistry. 2011; 166-182.
6. Cui JJ, et al. Structure based drug design of crizotinib, a potent and selective dual inhibitor of mesenchymal-epithelial transition factor kinase and anaplastic lymphoma kinase. J Med Chem. 2011;54(18):6342–6363.
7. Huggins DJ, et al. Rational approaches to improving selectivity in drug design. J Med Chem. 2012;55(4):1424-44.
8. Yap DB, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451-2459.
9. Mooij WT, et al. ProteinCCD: enabling the design of protein truncation constructs for expression and crystallization experiments. Nucleic Acids Research. 2009;37:W402–W405.
10. Gao X, et al. High-throughput limited proteolysis/mass spectrometry for protein domain elucidation. J Struct Funct Genomics. 2005;6(2-3):129-34.
11. Savva R, et al. DNA fragmentation based combinatorial approaches to soluble protein expression Part II: library expression, screening and scale-up. Drug Discov Today.
12. Savva R, et al. DNA fragmentation-based combinatorial approaches to soluble protein expression Part I. Generating DNA fragment libraries. Drug Discov Today.
13. Meier C, et al. Engineering human MEK-1 for structural studies: A case study of combinatorial domain hunting. J Struct Biol. 2012;177(2):329-34.
14. Reich S, et al. Combinatorial Domain Hunting: An effective approach for the identification of soluble protein domains adaptable to high-throughput applications. Protein Sci. 2006;15(10):2356–2365.
15. Laing VE, et al. Fused thiophene derivatives as MEK inhibitors. Bioorg Med Chem Lett. 2012;22(1):472-5.
16. Maclagan K, et al. A combinatorial method to enable detailed investigation of protein-protein interactions. Future Med Chem. 2011;3(3):271-82
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








