

 About Authors: Mrugank BP1, Mangala L2, Hareesha RP3
About Authors: Mrugank BP1, Mangala L2, Hareesha RP3
Department(s) and institution(s)
1. National Institute of Pharmaceutical Education and Research (NIPER), Department of Pharmacy Practice, M.Pharm (Student)
2. Gauhati Medical College and Hospital (GMCH), Department of Pharmacology, Prof and Head; Chief academic co-ordinator, NIPER-Ghy.
3. National Institute of Pharmaceutical Education and Research (NIPER), Department of Pharmacy Practice, M.Pharm (Student)
Reference ID: PHARMATUTOR-ART-1048
Abstract:
Background: "Pharmacovigilance is the science and activity relating the detection, assessment, understanding and prevention of adverse effects or any other possible drug - related problems." [1]
Literature:Periodic safety update reports (PSUR)[2]and Medical Dictionary for Regulatory Activities (MedDRA)[3]are two important assets for maintenance of Drug safety Pharmacovigilancesystem as they are designed to represent the safety data on a particular drug from all the sources and geographical regions in the worldand to understand internationally, accepted clinically validated medical terminology for medical coding respectively.
[adsense:336x280:8701650588]
Finding:Indian market has mostly seen the launch of only those products that have been already approved and marketed in the regulated markets of USA, Europe, Japan or other countries. For assessing the benefit-risk profile of a drug and to take appropriate corrective actions, the Indian pharmaceutical companies as well as the regulators have been depending on the experiences gained from these markets where the drug was being used for several years before introduction in India, thus bypassing the requirement to establish a strong Pharmacovigilance system of their own.[4]
Key message: India is becoming a major hub for clinical research activities due to its large population and well-defined endogamous subpopulations,[5]high enrollment rates and low costs. Moreover, with more than 300 medical colleges, 230 dental colleges, 830 pharmacy colleges and 657 recognized nursing colleges, there is a great opportunity for India to tap the potential human resources required for an effective Pharmacovigilance system.[6]
Introduction:
Why Pharmacovigilance system in INDIA is on Growth?
The Pharmaceutical industry in India is valued at Rs. 90,000 Crore and is growing at the rate of 12 – 14 % per annum. Exports are growing at 25 % Compound Annual Growth Rate (CAGR) every year. The total export of Pharma products is to the extent of Rs. 40,000 Crore. India is now being recognized as the ‘Global pharmacy of Generic Drugs’ & has distinction of providing generic quality drugs at affordable cost. India is also emerging rapidly as a hub of Global Clinical trials & a destination for Drug Discovery & Development.[7]
Further, more & more new drugs are being introduced into the country which include New Chemical Entities (NCE), high tech pharma products, vaccines as well as new dosage forms, new routes of drug administrations and new therapeutic claims of existing drugs. This is reflected in the fact that total numbers of applications received and processed have more than doubled from around 10,000 in the Year 2005 to 22,806 in Year 2009 at CDSCO, HQ, New Delhi. This includes increase in New Drug Applications, Global Clinical Trials, Market Authorization of Vaccine & Biotech products from 1200,100, 10 in Year 2005 to 1753, 262 & 137 in the Year 2009 respectively.[7]
Such rapid induction of NCEs and high tech Pharma products in the market throw up the challenges of monitoring Adverse Drug Reactions (ADRs) over large population base.
All medicines (pharmaceuticals and vaccines) have side effects. Some of these side effects are known, while many are still unknown even though that medicine has been in clinical use for several years. It is important to monitor both the known and hitherto unknown side effects of medicines in order to determine any new information available in relation to their safety profile. In a vast country like India with a population of over 1.2 Billion with vast ethnic variability, different disease prevalence patterns, practice of different systems of medicines, different socioeconomic status, it is important to have a standardized and robust Pharmacovigilance and drug safety monitoring programme for the nation. Collecting this information in a systematic manner and analyzing the data to reach a meaningful conclusion on the continued use of these medicines is the rationale to institute this program for India.[6]
Since, there are considerable social and economic consequences of ADRs there is a need to engage health-care professionals, in a well structured programme to build synergies for monitoring ADRs. The purpose of the Pharmacovigilance Program of India is to collect, collate and analyze data to arrive at an inference to recommend regulatory interventions, besides communicating risks to healthcare professionals and the public.
Target for coming five years Pharmacovigilance Programme of INDIA (June 2010- March 2015)launched by CDSCO
The Central Drugs Standard Control Organization (CDSCO), Directorate General of Health Services under the aegis of Ministry of Health & Family Welfare, Government of India in collaboration with Department of Pharmacology, All India Institute of Medical Sciences (AIIMS), New Delhi has launched the nation-wide Pharmacovigilance programme for protecting the health of the patients by assuring drug safety. The programme is coordinated by the Department of Pharmacology at AIIMS as a National Coordinating Centre (NCC).[7]Roadmap & detailed target phases of programme is shown in Fig. 1 & Fig. 2 respectively as below.[7]
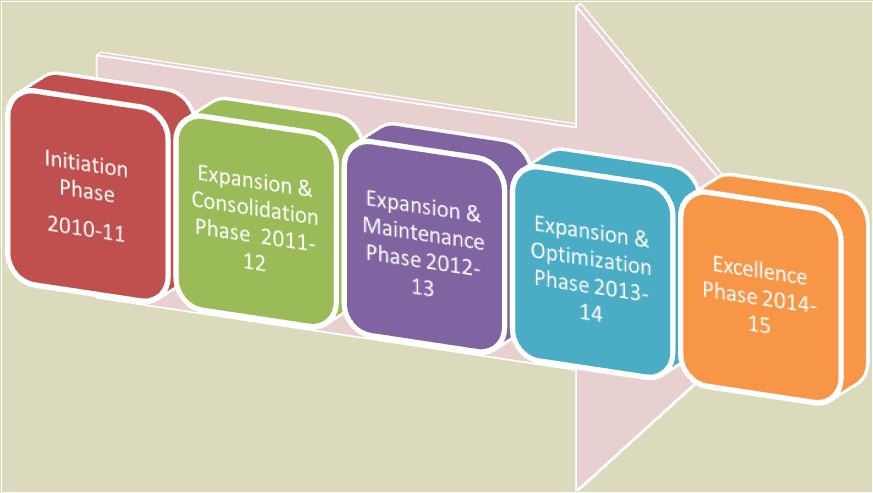
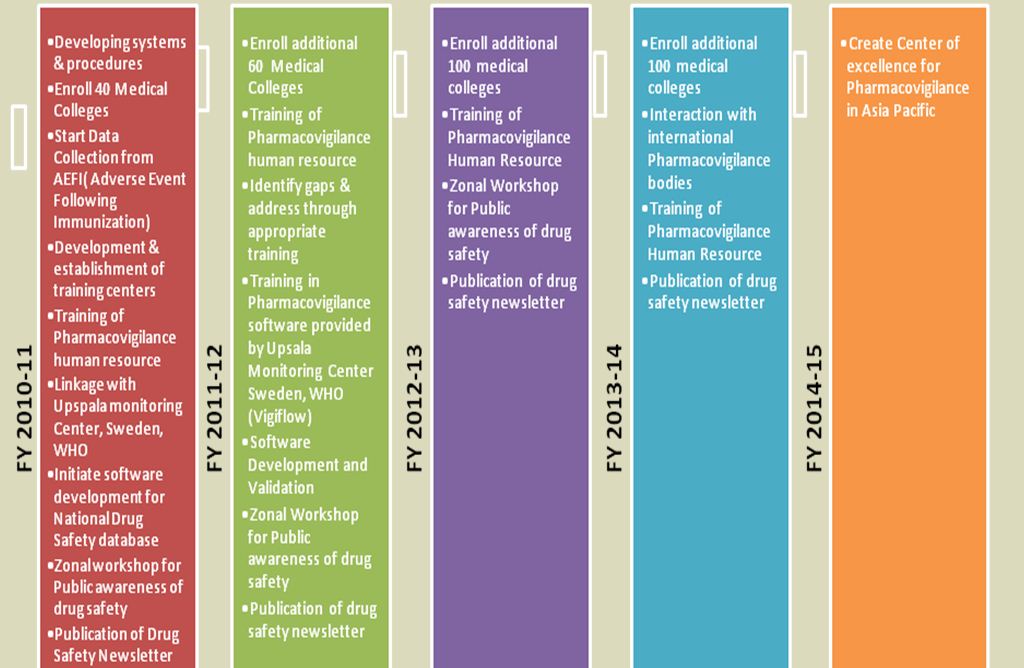
Literature:
PSURs (Periodic Safety Update Reports)
PSUR is a first important tool as Assets of Drug safety Pharmacovigilance system. PSURhave been designed to provide the regulators with an update of the worldwide safety data of a marketed drug, biological product or device at defined time intervals.[2] It is considered to be an important pharmacovigilance tool as it is designed to include the safety data on a particular drug from all the sources and geographical regions. Like other regulators, Drug Controller General of India (DCGI) also recommends a single PSUR for all dosage forms, formulations, and indications for one active substance. Within a single PSUR, data for different dosage forms, indications or populations should be provided separately. License holders are expected to include succinct summary information along with the critical evaluation of the safety profile of a marketed drug in the light of new changes during post-authorization period. A PSUR should also mention whether further investigations need to be carried out and what changes need to be made in the package insert.
Format of PSURs
Format of PSUR provided in the Schedule Y is similar to that of ICH E2C [2] format, although it does not elaborately guide the contents of the data to be incorporated under each and every heading. For all practical purposes, a PSUR prepared in accordance with ICH E2C format should be acceptable to DCGI.
PSUR reporting cycle
The current situation for periodic safety reports on marketed drugs is different among the three ICH regions.[2]
Schedule Y recommends that for all new products, PSURs should be submitted every 6 months for the initial 2 years and thereafter annually for the next 2 years. It is quite similar to the reporting cycle requirements of European Union (EU).
- In the EU, Council Directive 93/39/EEC and Council Regulation 2309/93 require reports with a periodicity of 6 months for two years, annually for the three following years and then every five years, at time of renewal of registration.
Where it is quite differed from US FDA periodic safety reporting system.
- The US regulations require quarterly reports during the first 3 years, then annual reports. The FDA has recently published proposed rules [8] which take into account the CIOMS Working Group II proposals [9].
- In Japan, the authorities require a survey on a cohort of a few thousand patients established by a certain number of identified institutions during the 6 years following authorization. Systematic information on this cohort, taking into account a precise denominator, must be reported annually. Regarding other marketing experience, adverse reactions which are non-serious, but both mild in severity and unlabeled must be reported every 6 months for 3 years and annually thereafter.
Like other regulatory authorities, DCGI can also extend the total duration for the submission of PSURs if it is considered necessary in the interest of public health. Like other major regulatory authorities, Indian regulations also require that PSURs should contain the relevant clinical and non-clinical safety data only for the period of report (interval data). Although it is not specified in Schedule Y, as per ICH E2C requirements, PSURs submitted to DCGI contain cumulative data on the regulatory status information on authorization applications and renewals, as well as data on serious, unlisted adverse reactions. Periodic safety update reports due for a period must be submitted within 30 calendar days for the last day of the reporting period. As there is no guidance available on the data lock, generally pharmaceutical companies follow the recommendations from ICH E2C for the data lock.
Period of reporting in PSURs
Schedule Y further states that if the marketing of a new drug is delayed after obtaining the approval to market, such data may be submitted on a deferred basis beginning from the time the new drug is marketed. This is in sharp contrast to EU regulations, where a pharmaceutical company is required to meet pharmacovigilance obligations of all the products for which it holds marketing authorization, irrespective of the marketing status of the products. As there is limited guidance available in Schedule Y as well as the protocol published by the NPP, it is very important for the pharmacovigilance team of an Indian pharmaceutical company to consult the guidance documents available from ICH, US FDA, and EMEA so as to develop well laid down
procedures for optimally carrying out the pharmacovigilance of new as well as generic drugs. Some additional activities that are generally carried out by a generic pharmaceutical company, however, have not been covered above include literature searches, generation of alerts, communications to healthcare professionals, and execution of pharmacovigilance agreements.
MedDRA (Medical Dictionary for Regulatory Activities)
What is MedDRA?
MedDRAis a second most important tool as Assets of Drug safety Pharmacovigilance system.
In 1997 the first version of MedDRA was born.[10]
It was developed from the European Union system, under the auspices of the ICH (International Conference on Harmonization).
It was developed to standardise regulatory communication between the authorities responsible for the authorization of medicinal products as well as the exchange between authorities and biopharmaceutical companies; data related to safety, quality or efficacy of medicinal products.
MedDRAhas been developed for “Medical Coding” as an internationally, clinically validated medical terminology for utilization in data entry, retrieval, evaluation and presentation, in both pre-and post marketing phases of regulatory processes.
Why knowledge of MedDRA is Important?
The coding of patient data is critical in the grouping, analysis, and reporting of data. Coding decisions directly impact submissions for New Drug Applications (NDAs), safety surveillance, and product labeling.[10]The success of a submission to the FDA can be significantly impacted by the analysis of adverse events, medical history and concomitant medications. The analysis relies on the interpretation of what has been transcribed from the subject CRF (Case Report Form).[10]
The original clinical term is referred to as the clinician’s term or verbatim term. This term needs to be re-interpreted or coded into a preferred term in order for it to be used during analysis. This is because different verbatim terms can have the same meaning such as in the example of the term “pain in head” or “headache”. In this case, the two distinct verbatim terms are coded to one synonymous preferred term. The identical terms and the consistent classification of the term allow the analysis to draw valid statistical conclusions pertaining to the subject’s experience. The coding process can therefore affect the statistical interpretation of the adverse events or medications in which the subject is taking during the clinical trial.[10]
Structure of MedDRA:
MedDRA is a five level multi-axial terminology. [11] The relationships between terms fall into one of three categories:
Hierarchical - provides vertical links between superordinate (broad grouping) terms and subordinate (higher level of specificity) descriptors.
1. System Organ Class (SOC)
2. High Level Group Term (HLGT)
3. High Level Term (HLT)
4. Preferred Term (PT)
5. Lowest Level Term (LLT)
Equivalence - grouping of synonymous terms. This is only exemplified in the relationship between the LLT and the PT.
Associative - allows terms to be linked horizontally, which are neither equivalent, nor hierarchically related but have a strong relation by sign, symptom, disease and diagnosis. This in only exemplified in the Special Search Categories.
The 26 System Organ Classes (SOC) in MedDRA represents parallel axes, which are not mutually exclusive. This allows terms to be represented in more than one SOC, and therefore grouped by different classifications. One single medical concept can be represented in more than one medical discipline. For example, the term Congenital HIV infection is represented in the following SOCs:
• Congenital and familial/genetic disorders
• Pregnancy, puerperium and perinatal conditions
• Infections and infestations
• Immune system disorders
Scope & Purpose of using MedDRA:
Terms found in MedDRA [11]
• Diseases
• Diagnosis
• Signs & Symptoms
• Therapeutic indications
• Investigation names and qualitative results
• Medical and surgical procedures
• Medical, social, family history
• Terms from: COSTART, WHO-ART, ICD-9 (some), ICD-9CM and HARTS.
Terms NOT found in MedDRA [11]
• Population level qualifiers (e.g. rare and frequent fail to focus on the individual patient)
• Numerical values for results (you cannot universalise numeric representations, especially in terms of the measurement parameter)
• Severity descriptors (typically, terms such as severe or mild are not found in the terminology, some exception when their presence is medically relevant, e.g. aggravated conditions are different than the condition its self)
• Medical product terms (except a very small number pertaining to clinical lab tests)
• Patient demographics (aside from very few occasions where sex is a pertinent descriptor, terms like age, race and religion are not included in the terminology
• Equipment, device and diagnostic product terms (e.g. the term catheter would not be include in the terminology where as the failure and its health effects would be)
Purposes of Using MedDRA [12]
• To aggregate reported terms in medically meaningful groupings for the purpose of reviewing and/or analyzing safety data.
• To facilitate identification of common data sets for evaluation of clinical and safety information.
• To facilitate consistent retrieval of specific cases or medical conditions from a database.
• To improve consistency in comparing and understanding “safety signals” and aggregated clinical data.
• To facilitate electronic data interchange of clinical safety information.
• To report adverse reaction/adverse event (ADR/AE) terms via individual case safety reports.
• To include ADR/AEs in tables, analyses, and line listings for reports
• To identify frequency of medically similar ADR/AEs.
• To capture and present product indications, investigations, medical history and social history data.
Conclusion:
• The lag period between when a drug is placed for the first time on the market in the United States, Europe, Japan or somewhere else in the world and its subsequent availability in India has decreased considerably. As a result for such drugs, the long-term safety data is not available at the time of their marketing in India or is otherwise significantly reduced. [4] In such cases, the Indian regulatory agencies cannot count on the experience of other markets to assess the benefit-risk balance of a drug—a fact that underscores the need to develop and implement an adequately designed pharmacovigilance system in India.
• This paper presents the clinical significance of coding decisions and how it affects your analysis. This has an impact on your submission resulting in the failure or success of a FDA filing.
• Indeed, for any pharmacovigilance system to operate well, knowledge of PSURs and MedDRA playing an ornamental role.
• All of these factors have drawn the attention of not only DCGI but also WHO and pharmaceutical companies – that INDIA has capabilities to do so and so all of which have to play an important role in the ultimate functioning of Pharmacovigilance activities in India.
Reference:
1. World Health Organization (WHO) (A). The Importance on Pharmacovigilance. Safety Monitoring on Medicinal Products. Geneva (Switzerland): Office of Publications, World Health Organization; 2002.
2. ICH E2C (R1): clinical safety data management: periodic safety update reports for marketed drugs.
3. Volume 9 of Pharmacovigilance, The rules governing medicinal products in the European Union.
4. Nair MD. Pharmacovigilance: the need for a formal system in India, 2001.
5. “Genetic jackpot in India’s endogamous gene pool,” IndiaInfo.com, June 13, 2005.
6. Y. K. Gupta, Ensuring Patient Safety - Launching the New Pharmacovigilance Programme of India, Pharma Times.2010;42(8):21-26.
7. Pharmacovigilance Programme of India (PvPI) for Assuring Drug Safety, Sponsored and coordinated by CDSCO, in collaboration with Department of Pharmacology, All India Institute of Medical Sciences (AIIMS), New Delhi, 2010.
8. Adverse Experience Reporting Requirements for Human Drug and Licensed Biological Products; Proposed Rule, Federal Register, 27 October 1994, pp. 54046-540
9. International Reporting of Periodic Drug-Safety Update Summaries. Final Report of CIOMS Working Group II. CIOMS - Geneva 1992
10. Sy Truong et al. Can Coding MedDRA and WHO Drug be as easy as a Google Search? SAS Global Forum.2007, paper 158.
11. MedDRA Maintenance and Support Services Organization (MSSO).available at meddramsso.com
12. MedDRA TERM SELECTION: POINTS TO CONSIDER, Release 3.2 Based on MedDRA version 6.1, ICH-Endorsed Guide for MedDRA Users, 18 July 2003.
Find More Articles at our portal






