
Presently, the name tetracycline refers to a number of antibiotics of either natural, or semi-synthetic origin, derived from a system of four linearly annelated six-membered rings (1,4,4a,5,5a,6,11,12a-octahydronaphthacene) with a characteristic arrangement of double bonds. The tetracycline molecule possesses five asymmetric centers: C-4, -4a, -5a, -6, and -12a.
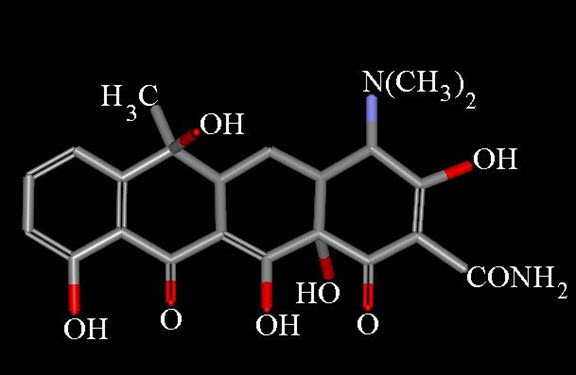
Chemical properties
The reactions that tetracyclines undergo are generally of a sophisticated nature, dictated by the complex functionality and the sensitivity of the molecules to mild reaction conditions (acid, base, heat).
Acidic conditions
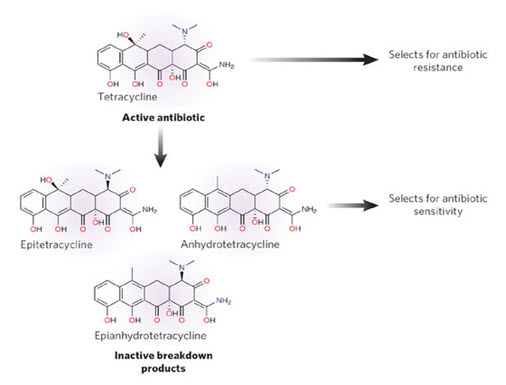
When exposed to dilute acid conditions, tetracycline undergoes dehydration to yield anhydrotetracycline. Anhydroterramycin suffers further cleavage and lactonization to apoterramycin:
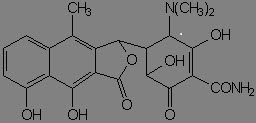
Diluted acid promotes epimerization at C-4 as well.
Basic conditions
Mild alkali attacks 11a carbon of tetracycline, which is transformed to isotetracycline:
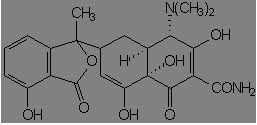
Formation of complexes
Tetracycline possesses a great tendency to form complexes with a number of chemical species, due to its B- and C-ring oxygen atoms:
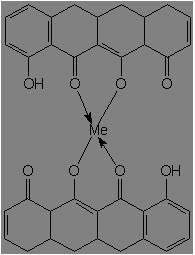
It complexes most readily with Fe3+, Fe2+, Cu2+, Ni2+, Co2+, Zn2+, Mn2+, Mg2+, Ca2+, Be2+, Al3+ among metal ions, phosphates, citrates, salicylates, p-hydroxybenzoates, saccharin anion, caffiene, urea, thiourea, polivinylpyrrolidone, serum albumin, lipoproteins, globulins, and RNA
MECHANISM OF ACTION
Tetracycline is a short-acting antibiotic that inhibits bacterial growth by inhibiting translation. It binds to the 30S ribosomal subunit and prevents the amino-acyl tRNA from binding to the A site of the ribosome. It also binds to some extent to the 50S ribosomal subunit. This binding is reversible in nature. Additionally tetracycline may alter the cytoplasmic membrane of bacteria causing leakage of intracellular contents, such as nucleotides, from the cell.
Pharmcokinetics:
Metabolism:
Absorption: Bioavailability is less than 40% when administered via intramuscular injection, 100% intravenously, and 60-80% orally (fasting adults). Food and/or milk reduce GI absorption of oral preparations of tetracycline by 50% or more.
Protein binding: 20 - 67% protein bound
Route of elimination They are concentrated by the liver in the bile and excreted in the urine and feces at high concentrations in a biologically active form. Half life is 6-12 hours.
Antimicrobial properties
In the 1950s, when most of the tetracyclines were discovered, their antimicrobial spectrum was broader than of any othen antibiotic then known. Tetracyclines are characterized by their exceptional chemotherapeutic efficacy against a wide range of Gram positive and Gram negative bacteria, richettsia, spirochetes, and large viruses, such as members of the lymphogranuloma group. The main indications for the use of tetracyclines are infections due to Escherichia coli and Haemophilus influenzae, infections of the bile duct, bacterial respiratory disorders including bronchitis prophylaxis, mixed infections arising from the mouth, pharynx, or intestinal tract, brucellosis, tularemia, plague and other pasteurelloses, leptospirosis, lymphogranuloma inguinale, cholera, and rickettsiosis. Because of the development of strains of microorganisms resistant to the tetracyclines, these antibiotics have lost some of their usefulness. They are no longer the drugs of first choice for treatment of staphylococcal, streptococcal, or pneumococcal infections. The individual tetracyclines differ less in their potency that in pharmacokinetic properties such as resorption, tissue diffusion, and elimination.
RESISTANCE: Two genes have been recognized that contribute to the resistance in tetracyclines tet and otr . They donot allow the binding of tetracycline with the aminoacyl t RNA by changing the adenine to guanine.
STRUCTURE-ACTIVITY RELATIONSHIPS
1. Tetracycline molecules comprise a linear fused tetracyclic nucleus (rings designated A, B, C, and D to which a variety of functional groups are attached. The simplest tetracycline to display detectable antibacterial activity is 6-deoxy-6-demethyltetracycline and so this structure may be regarded as the minimum pharmacophore.
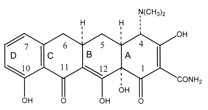
2. Features important for antibacterial activity among the tetracyclines are maintenance of the linear fused tetracycle, naturally occurring (α) stereochemical configurations at the 4a, 12a (A-B ring junction), and 4 (dimethylamino group) positions, and conservation of the keto-enol system (positions 11, 12, and 12a) in proximity to the phenolic D ring.
3. Chelation sites include the β-diketone system (positions 11 and 12) and the enol (positions 1 and 3) and carboxamide (position 2) groups of the A ring.
4. Replacement of the C-2 carboxamide moiety with other groups has generally resulted in analogs with inferior antibacterial activity, probably because bacteria accumulate these molecules poorly.
5. However, the addition of substituents to the amide nitrogen can impart significant water solubility, as in the case of rolitetracycline and lymecycline.
6. Consistent with the above observations, substitutions at positions 1, 3, 4a, 10, 11, or 12 are invariably detrimental for antibacterial activity.
7. A number of other substitutions at different positions on the B, C, and D rings are tolerated, and molecules possessing these substituents have given rise to the tetracyclines in clinical use today, as well as the new glycylcycline molecules that are currently undergoing clinical trials.
8. The extensive structure-activity studies referred to above revealed that with one exception, each of the rings in the linear fused tetracyclic nucleus must be six membered and purely carbocyclic for the molecules to retain antibacterial activity.
9. For instance, the nortetracyclines, derivatives in which the B ring comprises a five-membered carbocycle, are essentially devoid of antibacterial activity . Nevertheless, 6-thiatetracycline, which possesses a sulfur atom at position 6 of the C ring, is an apparent exception to the rule that a purely carbocyclic six-membered ring structure is required for activity, since molecules in this series have potent antibacterial properties.
Atypical tetracyclines:
These molecules, which also include the anhydrotetracyclines, 4-epi-anhydrotetracyclines, and chelocardin, appear to directly perturb the bacterial cytoplasmic membrane, leading to a bactericidal response (43, 198, 199). This contrasts with the typical tetracyclines, which interact with the ribosome to inhibit bacterial protein synthesis and display a reversible bacteriostatic effect. The membrane-disrupting properties of the atypical tetracyclines are probably related to the relative planarity of the B, C, and D rings so that a lipophilic, nonionized molecule predominates. On interaction with the cell, the atypical tetracyclines are likely to be preferentially trapped in the hydrophobic environment of the cytoplasmic membrane, disrupting its function. These molecules are of no interest as therapeutic candidates because they cause adverse side effects in humans (250), which are probably related to their ability to interact nonspecifically with eukaryotic as well as prokaryotic cell membranes.
Synthesis:
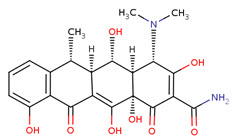
Doxycycline:
A synthetic tetracycline derivative with similar antimicrobial activity. Animal studies suggest that it may cause less tooth staining than other tetracyclines
Mecahnism:
Doxycycline, is lipophilic and can pass through the lipid bilayer of bacteria. Doxycycline reversibly binds to the 30 S ribosomal subunits and possibly the 50S ribosomal subunit(s), blocking the binding of aminoacyl tRNA to the mRNA and inhibiting bacterial protein synthesis. Doxycycline prevents the normal function of the apicoplast of Plasmodium falciparum, a malaria causing organism.
Pharmacokinetics:
Completely absorbed following oral administration. >90%Protein binding, undergoes hepatic metabolism. Route of elimination is it is concentrated by the liver in the bile and excreted in the urine and feces at high concentrations in a biologically active form. Half life is 18-22 hours.
Uses:
Doxycycline is indicated for use in respiratory tract infections caused by Mycoplasma pneumoniae, Haemophilus influenzae, Streptococcus pneumoniae, Legionella spp., or Klebsiella spp. It is also used for prophylaxis of malaria. Doxycycline is indicated for a variety of bacterial infections, from Mycobacterium fortuitum and M. marinum, to susceptible E. coli and Brucella spp. It can be used as an alternative to treating plague, tetanus, Campylobacter fetus.
Toxicity:
Symptoms of overdose include anorexia, nausea, diarrhoea, glossitis, dysphagia, enterocolitis and inflammatory lesions (with monilial overgrowth) in the anogenital region, skin reactions such as maculopapular and erythematous rashes, exfoliative dermatitis, photosensitivity, hypersensitivity reactions such as urticaria, angioneurotic oedema, anaphylaxis, anaphyl-actoid purpura, pericarditis, and exacerbation of systemic lupus erythematosus, benign intracranial hypertension in adults disappearing on discontinuation of the medicine, haematologic abnormalities such as haemolytic anaemia, thrombocytopenia, neutropenia, and eosinophilia.
Oxytetracycline
A tetracycline analog isolated from the actinomycete Streptomyces rimosus and used in a wide variety of clinical conditions.
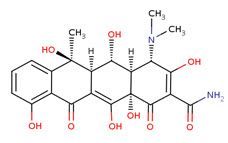
Pharmacodynamics:
Oxytetracycline is known as a broad-spectrum antibiotic due to its activity against such a wide range of infections. It was the second of the tetracyclines to be discovered. Oxytetracycline, like other tetracyclines, is used to treat many infections common and rare. Its better absorption profile makes it preferable to tetracycline for moderately severe acne, but alternatives sould be sought if no improvement occurs by 3 months.
Oxytetracycline inhibits cell growth by inhibiting translation. It binds to the 30S ribosomal subunit and prevents the amino-acyl tRNA from binding to the A site of the ribosome. The binding is reversible in nature. Oxytetracycline is lipophilic and can easily pass through the cell membrane or passively diffuses through porin channels in the bacterial membrane.
Metabolism:
Indication
Oxytetracycline is indicated for treatment of infections caused by a variety of Gram positive and Gram negative microorganisms including Mycoplasma pneumoniae, Pasteurella pestis, Escherichia coli, Haemophilus influenzae (respiratory infections), and Diplococcus pneumoniae.
Toxicity
Adverse effects may include stomach or bowel upsets and rarely allergic reactions. Very rarely severe headache and vision problems may be signs of dangerous intracranial hypertenion.
Minocycline
A tetracycline analog, having a 7-dimethylamino and lacking the 5 methyl and hydroxyl groups, which is effective against tetracycline-resistant staphylococcus infections.
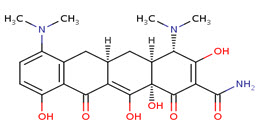
Minocycline, the most lipid soluble and most active tetracycline antibiotic, is, like doxycycline, a long-acting tetracycline. Minocycline's effects are related to the inhibition of protein synthesis. Although minocycline's broader spectrum of activity, compared to other members of the group, includes activity against Neisseria meningitidis, its use as a prophylaxis is no longer recomended because of side effects (dizziness and vertigo). Current research is examining the possible neuroprotective effects of minocycline against progression of Huntington's Disease, an inherited neurodegenerative disorder. The neuroprotective action of minocycline may include its inhibitory effect on 5-lipoxygenase, an inflammatory enzyme associated with brain aging.
Mechanism of action
Minocycline passes directly through the lipid bilayer or passively diffuses through porin channels in the bacterial membrane. Tetracyclines like minocycline bind to the 30S ribosomal subunit, preventing the binding of tRNA to the mRNA-ribosome complex and interfering with protein synthesis.
Pharmacokinetics:
Rapidly absorbed from the gastrointestinal tract and absorption is not significantly impaired by ingestion of food or milk. Oral bioavailability is 100%. 55% to 76% protein binding. Undergoes hepatic metabolism. Half life is 11-22 hours.
Indication
For the treatment of infections caused by susceptible strains of microorganisms, such as Rocky Mountain spotted fever, typhus fever and the typhus group, Q fever, rickettsial pox and tick fevers caused by Rickettsiae, upper respiratory tract infections caused by Streptococcus pneumoniae and for the treatment of asymptomatic carriers of Neisseria meningitidis.
Toxicity
Minocycline has been observed to cause a dark discoloration of the thyroid in experimental animals (rats, minipigs, dogs and monkeys). In the rat, chronic treatment with minocycline has resulted in goiter accompanied by elevated radioactive iodine uptake and evidence of thyroid tumor production.
Lymecycline
A tetracycline with a 7-chloro substitution.
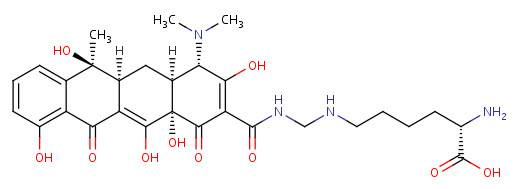
Pharmacodynamics
Lymecycline is a tetracycline broad-spectrum antibiotic. It is approximately 5000 times more soluble than tetracycline base and is unique amongst tetracyclines in that it is absorbed by the "active transport" process across the intestinal wall, making use of the same fast and efficient mechanism by which carbohydrates are absorbed. It inhibits cell growth by inhibiting translation.
Lymecycline inhibits cell growth by inhibiting translation. It binds to the 30S ribosomal subunit and prevents the amino-acyl tRNA from binding to the A site of the ribosome. The binding is reversible in nature. Lymecycline is lipophilic and can easily pass through the cell membrane or passively diffuses through porin channels in the bacterial membrane. Cells become resistant to lymecycline by at least two mechanisms: efflux and ribosomal protection. In efflux, a resistance gene encodes a membrane protein that actively pumps lymecycline out of the cell. This is the mechanism of action of the tetracycline resistance gene on the artificial plasmid pBR322. In ribosomal protection, a resistance gene encodes a protein which binds to the ribosome and prevents lymecycline from acting on the ribosome.
Pharmacokinetics:
Absorption is fast and efficient. Bioavailability is 100% following oral administration.
Indication
For the treatment of infections and to treat acne. It may also be used to treat urinary tract infections, gum disease, and other bacterial infections such as gonorrhea and chlamydia. Lymecycline is also used commonly as a prophylactic treatment for infection by Bacillus anthracis (anthrax). It is also effective against Yersinia pestis and malaria and is also prescribed for the treatment of Lyme disease.
Toxicity
Adverse effects include nausea, vomiting, diarrhoea, glossitis, enterocolitis, dysphagia, dermatitis, hypersensitivity reactions, proctitis, and vaginitis.
Rolitetracycline
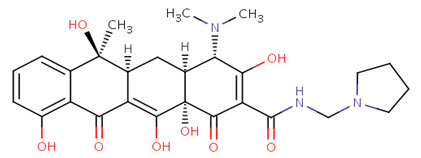
Mechanism of action
Rolitetracycline is a semisynthetic broad-spectrum tetracycline antibiotic used especially for parenteral administration in cases requiring high concentrations or when oral administration is impractical. Rolitetracycline passively diffuses through porin channels in the bacterial membrane and reversibly binds to the 30S ribosomal subunit, preventing binding of tRNA to the mRNA-ribosome complex, and thus interfering with protein synthesis.
Indication
Rolitetracycline is a broad-spectrum antibiotic used in cases needing high concentrations or when oral administration is impractical.
Toxicity
Symptoms of overdose include anorexia, nausea, diarrhoea, glossitis, dysphagia, enterocolitis and inflammatory lesions (with monilial overgrowth) in the anogenital region, skin reactions such as maculopapular and erythematous rashes, exfoliative dermatitis, photosensitivity, hypersensitivity reactions such as urticaria, angioneurotic oedema, anaphylaxis, anaphyl-actoid purpura, pericarditis, and exacerbation of systemic lupus erythematosus, benign intracranial hypertension in adults disappearing on discontinuation of the medicine, haematologic abnormalities such as haemolytic anaemia, thrombocytopenia, neutropenia, and eosinophilia.
Tigecycline
Tigecycline is a glycylcycline antibiotic developed and marketed by Wyeth under the brand name Tygacil. It was given a U.S. Food and Drug Administration (FDA) fast-track approval and was approved on June 17, 2005. It was developed in response to the growing prevalence of antibiotic resistance in bacteria such as Staphylococcus aureus.
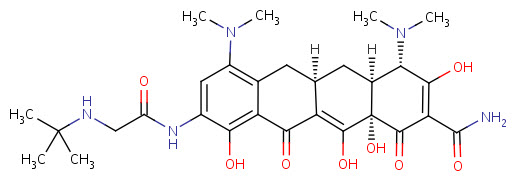
Tigecycline is the first clinically-available drug in a new class of antibiotics called the glycylcyclines. Glycylcyclines are a new class of antibiotics derived from tetracycline. These tetracycline analogues are specifically designed to overcome two common mechanisms of tetracycline resistance, namely resistance mediated by acquired efflux pumps and/or ribosomal protection. Glycylcycline antibiotics have a similar mechanism of action as tetracycline antibiotics. Both classes of antibiotics bind to the 30S ribosomal subunit to prevent the amino-acyl tRNA from binding to the A site of the ribosome. However, the glycylcyclines appear to bind more effectively than the tetracyclines.
Tigecycline is the first clinically-available drug in a new class of antibiotics called the glycylcyclines. Glycylcyclines are a new class of antibiotics derived from tetracycline. These tetracycline analogues are specifically designed to overcome two common mechanisms of tetracycline resistance, namely resistance mediated by acquired efflux pumps and/or ribosomal protection. Glycylcycline antibiotics have a similar mechanism of action as tetracycline antibiotics. Both classes of antibiotics bind to the 30S ribosomal subunit to prevent the amino-acyl tRNA from binding to the A site of the ribosome. However, the glycylcyclines appear to bind more effectively than the tetracyclines.
Metabolism
Tigecycline is not extensively metabolized. In vitro studies with tigecycline using human liver microsomes, liver slices, and hepatocytes led to the formation of only trace amounts of metabolites. A glucuronide, an N-acetyl metabolite, and a tigecycline epimer (each at no more than 10% of the administered dose) are the primary metabolites.
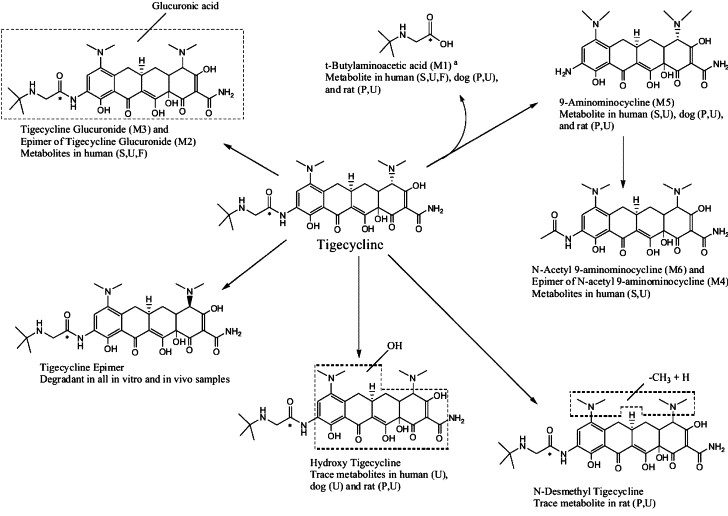
Indication
For the treatment of infections caused by susceptible strains of the designated microorganisms in the following conditions: Complicated skin and skin structure infections caused by Escherichia coli, Enterococcus faecalis (vancomycin-susceptible isolates only), Staphylococcus aureus (methicillin-susceptible and -resistant isolates), Streptococcus agalactiae, Streptococcus anginosus grp. (includes S. anginosus, S. intermedius, and S. constellatus), Streptococcus pyogenes and Bacteroides fragilis. Complicated intra-abdominal infections caused by Citrobacter freundii, Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Enterococcus faecalis (vancomycin-susceptible isolates only), Staphylococcus aureus (methicillin-susceptible isolates only), Streptococcus anginosus grp. (includes S. anginosus, S. intermedius, and S. constellatus), Bacteroides fragilis, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides vulgatus, Clostridium perfringens, and Peptostreptococcus micros.
Toxicity
Since glycylcyclines are similar to tetracyclines, they share many of the same side effects and contraindications as tetracyclines. These side effects may include nausea/vomiting, headache, photosensitivity, discoloration of growing teeth, and fetal damage.
--
ARTICLE BY,
Junaid Niazi
Asst Prof, Bahra Institute of Pharmacy,
Patiala
junaid468@gmail.com
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








