
About Author:Ratnadeep V. Ghadage.
Department of Pharmaceutical Chemistry,
Appasaheb Birnale College of Pharmacy, Sangli,
Maharashtra, India
ABSTRACT:
Any investigation in human subjects intended to discover or verify the clinical, pharmacological and/or other pharmacodynamic effects of an investigational product(s), and/or to identify any adverse reactions to an investigational product(s), and/or to study absorption, distribution, metabolism, and excretion of an investigational product(s) with the object of ascertaining its safety and/or efficacy. The terms clinical trial and clinical study are synonymous. Clinical trials examine the safety and efficacy of interventions, or treatments, in human subjects. This manuscript focuses on pharmaceutical clinical trials. The word subject is used deliberately here, since all participants in clinical trials are subjects, even if they are under the care of a personal physician, and therefore patients in that context, at the time of the trial. The pharmaceutical industry is now a more significant investor in clinical trials, but in addition to these opportunities, it is being challenged by the financial impact of managed care and medicaid regulations on academic medical-center revenues. Besides that, there is growing public concern about our systems for protection of human subjects, along with some conflicts. There are public expectations that clinical research will yield substantial advances in the health of the public. Once clinical research studies are completed and a drug has been approved for marketing by a regulatory agency, reports of the drug’s safety and efficacy will be published in the clinical literature. This dissemination of the results provides clinicians and research scientists with evidence of the beneficial administration of the drug.
[adsense:336x280:8701650588]
Refrence Id: PHARMATUTOR-ART-1147
INTRODUCTION 1,2:
Clinical trial is an inextricable link between advances in medical research technology and improved health care. It is a component of medical health research intended to produce knowledge valuable for understanding human disease, preventing and treating illness and promoting health. A clinical trial is the scientific term for a test or study of a drug, therapy, surgical procedure, medical device, or of nutrition or behavioral changes in people.
The tests are done to find out if the drug, therapy, procedure, etc. is safe and effective for people to use. The overall purpose of a clinical trial is to learn, not to treat patients. Before you volunteer to participate in a clinical trial, ask about any risks associated with the treatment being tested.
Objectives of Clinical trial:
- To assess the safety and effectiveness of a new medication or device on a specific kind of patients. (E.g. patients who have been diagnosed with Alzhiemer “s disease).
- To assess the safety and effectiveness of a different dose of a medication that is commonly used (e.g.10mg dose instead of 5mg dose).
- To assess the safety and effectiveness of an already marketed medication or devices for new indication.
- To assess whether the new medication or device is more effective for the patient’s condition than the already used, standard medication or device.
[adsense:468x15:2204050025]
Approval of clinical trials: Preclinical data obtained from animal’s studies provide a general pharmacological, toxicological and pharmacokinetic profile of the new drug.
These data are scrutinized by Expert Government Bodies in each country, with aim that clinical drug trials should be undertaken with great care and meticulous planning of the methodology adopted.
The New Drug Application (NDA) in the prescribed format, with all relevant literature and pre-clinical data must be submitted to the Drug ControlAuthority (DCA) for scrutiny, and sanction obtained before clinical evaluation studies are initiated.
Ethical data behind Clinical trials:3
Ethical Guidelines:
The goal of clinical research is to develop generalizable knowledge that improves human health or increases understanding of human biology. People who participate in clinical research make it possible to secure that knowledge. The path to finding out if a new drug or treatment is safe or effective, for example, is to test it on patient volunteers. But by placing some people at risk of harm for the good of others, clinical research has the potential to exploit patient volunteers. The purpose of ethical guidelines is both to protect patient volunteers and to preserve the integrity of the science.
- Clinical trials are closely supervised by appropriate regulatory authorities.
- All studies that involve a medical or therapeutic intervention on patients must be approved by a supervising ethics committee before permission is granted to run the trial.
Ethical Committee:
The sponsor or investigator should seek the opinion of an Independent Ethics Committee regarding suitability of the protocol, methods and documents to be used in recruitment of Subjects and obtaining their informed consent including adequacy of the information being provided to the Subjects. The Ethics Committees are entrusted not only with the initial view of the proposed research protocols prior to initiation of the projects but also have a continuing responsibility of regular monitoring for the compliance of the ethics of the approved programmes till the same are completed. Such an ongoing review is in accordance with the declaration of Helsinki and all the international guidelines for biomedical research.
Institution Review Board/Independent Ethics Committee (IRB/IEC): 5
It is an independent body constituted of medical professionals, scientific and non-scientific members, whose responsibility is to ensure the protection of the rights, safety and wellbeing of human subjects involved in a trial by among other things, reviewing, approving and providing continuing review of trial protocol and amendments and of the methods and material to be used in obtaining and documenting informed consent of the trial subjects.
The IECs should specify in writing the authority under which the Committee is established, membership requirements, the terms of reference, the conditions of appointment, the offices and the quorum requirements.
Procedure:
The IRB/IEC should establish, document in writing, and follow its procedures, which should include:
- Determining its composition (names and qualifications of the members) and the authority under which it is established.
- Scheduling, notifying its members of and conducting its meetings.
- Conducting initial and continuing review of trials.
- Determining the frequency of continuing review as appropriate.
- Providing, according to the applicable regulatory requirements, expedited review and approval or favourable opinion of minor change(s) in ongoing trials that have the approval or favourable opinion of the IRB/IEC.
- Specifying that no subject should be admitted to a trial before the IRB/IEC issues its written approval or favourable opinion of the trial.
- Specifying that no deviations from or changes of the protocol should be initiated without prior written IRB/IEC approval or favourable opinion of an appropriate amendment, except when necessary to eliminate immediate hazards to the subjects or when the change(s) involves only logistical.
-
Administrative aspects of the trial (e.g., change of monitor(s), telephone number(s)). Specifying that the investigator should promptly report to the IRB/IEC:
- Deviations from or changes of the protocol to eliminate immediate hazards to the trial subjects.
- Changes increasing the risk to subjects or affecting significantly the conduct of the trial.
- All adverse drug reactions (ADRs) that is both serious and unexpected.
- New information that may affect adversely the safety of the subjects or the conduct of the trial.
-
Ensuring that the IRB/IEC promptly notify in writing the investigator/institution concerning:
- Its trial-related decisions/opinions.
- The reasons for its decisions/opinions
ICH GCP: 4,5 Good clinical practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subject.
Role of FDA: The Food and Drug Administration is responsible for protecting the public health by assuring the safety, efficacy, and quality of drugs, biologics and many other medical products.The FDA is also responsible for advancing the public health by helping to speed innovations that make medicines more effective. Safer, and more affordable, and helping the public get the accurate, science-based information they need to medicines to improve their health.
However, FDA does not develop new therapies, or conduct the clinical trials to demonstrate safely and effectiveness. FDA staffs members meet with researchers, and perform in spections of clinical trials study sites to protect the rights of participants and to verify the quality and integrity of the data.
Clinical research organisation (CRO) [5: A CRO is an organisation to which the sponsor may transfer or delegate some or all of the tasks, duties and/ or obligations regarding a Clinical Study. All such contractual transfers of obligations should be defined in writing.
Schedule “Y” is gives requirements and guidelines for permission to import and /or manufacture of new drugs for sale or to undertake clinical trials.
Sponsor: It is an individual/ company, institution or organization which takes responsibility for the initiation, management, and financing of clinical trials.
A] Responsibilities of Sponsor:
a) The clinical trial Sponsor is responsible for implementing and maintaining quality assurance systems to ensure that the clinical trial is conducted and data generated, documented and reported in compliance with the protocol and Good Clinical Practice (GCP). Guidelines issued by the Central Drugs Standard Control Organization, Directorate General of Health Services, Government of India as well as with all applicable statutory provisions. Standard operating procedures should be documented to ensure compliance with GCP and applicable regulations.
b) Sponsors are required to submit a status report on the clinical trial to the Licensing Authority at the prescribed periodicity.
c) In case of studies prematurely discontinued for any reason including lack of commercial interest in pursuing the new drug application, a summary report should be submitted within 3 months. The summary report should provide a brief description of the study, the number of patients exposed to the drug, dose and duration of exposure, details of adverse drug reactions, if any, and the reason for discontinuation of the study or non-pursuit of the new drug application.
d) If any unexpected serious adverse event (SAE) occurring during a clinical trial should be communicated promptly (within 14 calendar days) by the Sponsor to the Licensing Authority and to the other Investigator(s) participating in the study.
B] Investigator and Institution Selection:The Sponsor is responsible for selecting the Investigator(s) / Institutions taking into account the appropriateness and availability of the study site and facilities. The Sponsor must assure itself of the Investigator’s qualifications and availability for the entire duration of the Study. If organisation of a co-ordinating committee and or selection of co-ordinating investigators are to be utilised in multi-centric studies their organisation and / or selection are Sponsor’s responsibilities.
C] Contract: The Sponsor should enter into a formal and legal agreement / contract with the Investigator(s) / Institution(s) on the following terms:
· To conduct the Study in compliance with GCP, the applicable regulatory requirements and the Protocol agreed to by the Sponsor and given approval / favourable opinion by the Ethics Committee.
· To comply with the procedures for data recording, and reporting.
· To permit monitoring, auditing and inspection.
· To retain the study related essential documents until the Sponsor informs the Investigator(s) / Institution(s) in writing that these documents are no longer needed.
· The agreement should define the relationship between the investigator and the sponsor in matters such as financial support, and payments in kind etc.
The Sponsor should establish detailed Standard Operating Procedures (SOP’s). The Sponsor and the Investigator(s) should sign a copy of the Protocol and the SOPs or an alternative document to confirm their agreement.
Compensation to Subject and Investigators:
- If required by the applicable regulatory requirement(s), the sponsor should provide insurance or should indemnify (legal and financial coverage) the investigator/the institution against claims arising from the trial, except for claims that arise from malpractice and/or negligence.
- The sponsor's policies and procedures should address the costs of treatment of trial subjects in the event of trial-related injuries in accordance with the applicable regulatory requirement(s).
Investigators: It is a person who is responsible for the conduct of the clinical trial at a trial site. If a trial is conducted by a team of individuals at a trial site, the investigator is the responsible leader of the team and may be called the principal investigator.
The investigator(s) should be qualified by education, training, and experience to assume responsibility for the proper conduct of the trial, should meet all the qualifications specified by the applicable regulatory requirement(s), and should provide evidence of such qualifications through up-to-date curriculum vitae and/or other relevant documentation requested by the sponsor, the IRB/IEC, and/or the regulatory authority.
- The investigator should be thoroughly familiar with the appropriate use of the investigational product(s), as described in the protocol, in the current Investigator's Brochure, in the product information and in other information sources provided by the sponsor.
- The investigator should be aware of, and should comply with, GCP and the applicable regulatory requirements.
- The investigator/institution should permit monitoring and auditing by the sponsor, and inspection by the appropriate regulatory authority (is).
- The investigator should maintain a list of appropriately qualified persons to whom the investigator has delegated significant trial-related duties.
Investigational Product: It is a pharmaceutical form of an active ingredient or placebo being tested or used as a reference in a clinical trial, including a product with a marketing authorization when used or assembled (formulated or packed) in a way different from the approved form, or when used for an unapproved indication, or when used to gain further information about the approved use.
Essential documents for the conduct of a clinical trial:
- Essential Documents are those documents which individually and collectively permit evaluation of the conduct of a trial and the quality of the data produced. These documents serve to demonstrate the compliance of the investigator, sponsor and monitor with the standards of Good Clinical Practice and with all applicable regulatory requirements.
- Essential Documents also serve a number of other important purposes. Filing essential documents at the investigator/institution and sponsor sites in a timely manner can greatly assist in the successful management of a trial by the investigator, sponsor and monitor. These documents are also the ones which are usually audited by the sponsor's independent audit function and inspected by the regulatory authority (is) as part of the process to confirm the validity of the trial conduct and the integrity of data collected.
- The minimum list of essential documents which has been developed follows. The various documents are grouped in three sections according to the stage of the trial during which they will normally be generated: 1) before the clinical phase of the trial commences, 2) during the clinical conduct of the trial, and 3) after completion or termination of the trial. A description is given of the purpose of each document, and whether it should be filed in either the investigator/institution or sponsor files, or both. It is acceptable to combine some of the documents, provided the individual elements are readily identifiable.
- Trial master files should be established at the beginning of the trial, both at the investigator/institution’s site and at the sponsor's office. A final close-out of a trial can only be done when the monitor has reviewed both investigator/institution and sponsor files and confirmed that all necessary documents are in the appropriate files.
Clinical trial Protocol 5,6:Clinical trial protocol is a document used to gain confirmation of the trial design by a panel of experts and adherence by all study investigators, even if conducted in various countries. It is a study plan on which all clinical trials are based. The plan is carefully designed to safeguard health of the participants as well as answer specific research questions.A protocol describes what types of people may participate in the trial, the schedule of tests procedures, medications, statistical consideration, organization of the planned trial and dosages, and the length of the study. The protocol contains a precise study plan for executing the clinical trial, not only to assure safety and health of the trial subjects, but also to provide an exact template for trial conduct by investigators at multiple locations(in a multicentre trial) to perform the study in exactly the someway. This harmonization allows data to be combined collectively as through all investigators were working closely together.
The protocol also give the study administrators as well as site team of physicians, nurses and clinical administrators a common reference document for site responsibility during the trial. The contents of a trial protocol should generally include the following topics
General Information:
- Protocol title, protocol identifying number, and date. Any amendment(s) should also bear the amendment number(s) and date(s).
- Name and address of the sponsor and monitor (if other than the sponsor).
- Name and title of the person(s) authorized to sign the protocol and the protocol amendment(s) for the sponsor.
- Name, title, address, and telephone number(s) of the sponsor's medical expert (or dentist when appropriate) for the trial.
- Name and title of the investigator(s) who is (are) responsible for conducting the trial, and the address and telephone number(s) of the trial site(s).
- Name, title, address, and telephone number(s) of the qualified physician (or dentist, if applicable), who is responsible for all trial-site related medical (or dental) decisions (if other than investigator). Name(s) and address (as) of the clinical laboratory (is) and other medical and/or technical department(s) and/or institutions involved in the trial.
Background Information:
i. Name and description of the investigational product(s).
ii. A summary of findings from nonclinical studies that potentially have clinical significance and from clinical trials that is relevant to the trial.
iii. Summary of the known and potential risks and benefits, if any, to human subjects.
iv. Description of and justification for the route of administration, dosage, dosage regimen, and treatment period(s).
v. A statement that the trial will be conducted in compliance with the protocol, GCP and the applicable regulatory requirement(s).
vi. Description of the population to be studied.
Clinical Trial Design6:
The scientific integrity of the trial and the credibility of the data from the trial depend substantially on the trial design. A description of the trial design should include:
i. A specific statement of the primary endpoints and the secondary endpoints, if any, to be measured during the trial.
ii. A description of the type/design of trial to be conducted (e.g. double-blind, placebo-controlled, parallel design) and a schematic diagram of trial design, procedures and stages.
iii. A description of the measures taken to minimize/avoid bias, including:
a. Randomization.
b. Blinding.
iv. A description of the trial treatment(s) and the dosage and dosage regimen of the investigational product(s). Also include a description of the dosage form, packaging, and labeling of the investigational product(s).
v. The expected duration of subject participation, and a description of the sequence and duration of all trial periods, including follow-up, if any.
vi. A description of the "stopping rules" or "discontinuation criteria" for individual subjects, parts of trial and entire trial.
vii. Accountability procedures for the investigational product(s), including the placebo(s) and comparator(s), if any.
viii. Maintenance of trial treatment randomization codes and procedures for breaking codes.
Types of clinical trials[5,6:
Clinical trials are used to study many aspects of health care:-
a. Prevention Trials:-It looks for better ways to prevent disease in people who have never had the disease or to prevent a disease from returning. These approaches may be including medicines, vitamins, vaccines, minerals, or lifestyle changes.
b. Screening Trials:-To test the best way to detect certain diseases or health conditions.
c. Diagnostic Trials:-Conduct to find better tests or procedures for diagnosing a particular disease or condition.
d. Treatment Trials:-To test experimental treatments, new combinations of drugs or new approaches to surgery or radiation therapy.
e. Quality of Trials:-It is to explore ways to improve comfort and quality of life for individuals with a chronic illness.
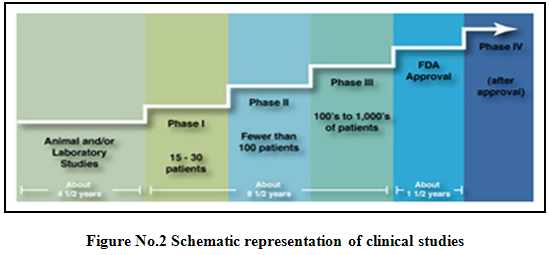
Phases of clinical trials5,6,9,10:
Clinical trials involving new drugs are commonly classified into four phases. The development process will normally proceed through all four phases over many years. If the drug successfully passes through phases I, II and III, it will usually approve.
Pre-clinical study:
Preclinical studies involve in vitro (test tube) and in vivo (animal) experiments using wide-ranging doses of the study drug to obtain preliminary efficacy, toxicity and pharmacokinetic information. Such tests assist pharmaceutical companies to decide whether a drug candidate has scientific merit for further development as an investigational new drug.
After a treatment is tested in the laboratory, it can go to human testing. There are four phases of human testing:
Phase I: - (Human pharmacology)
It is the process of testing an investigational drug on a smaller number of subjects to establish that it is safe for use in humans.
- Scope: A series of small tests to determine how healthy people (those without the condition or illness the drug will treat) react to and are affected by the treatment.
* Subjects: Healthy volunteers who are not taking other medicines.
* Number of volunteers: Less than 100.
* Duration: Several months.
There are different kinds of phase I trials:-
•Single Ascending Dose(SAD)
Single Ascending Dose studies are those in which small group of 3-6 patients receives
A small dose of the drug and observed for a specific period. If no adversed effects are observed, a new group of a patient is then given a higher dose,this is continued unit MTD (Maximum Tolerate Dose) is shown.
• Multiple Ascending Dose(MAD)
MAD studies, in this a group of patients receives a low dose of drug than dose is subsequently escalated and sample is collected at various times.It is conducted for better understanding of the pharmacokinetic and pharmacodynamic of the drug.
The clinical pharmacokineticist on a drug development team does not simply analyse data at the end of the study. Typically, he or she is intimately involved in the design of the study, either making or contributing to the decision regarding the first dose to be given to humans, the dose escalation scheme, and potentially the decision to escalate doses. Often the pharmacokineticist will have to justify the sensitivity limits for the bioanalytical methods proposed for measuring drug concentrations in human plasma. To facilitate this, a thorough understanding of preclinical models for activity (including target drug concentrations), ADME data, pharmacokinetics and toxicokinetics are required. This data can be characterized in the following ways:-
i] Useful Preclinical Pharmacology Data:
• Does the proposed pharmacological mechanism suggest that there should be a direct correlation between drug concentration and pharmacological effect, or is the effect more likely to be indirect?
• What is the IC50/EC50 needed to produce the desired effect?
• What is the IC5o (/EC50 that produces a toxic effect?
• Is there an in vitro/ex vivo correlation to the therapeutic or pharmacological effect?
• Is there a surrogate marker for the therapeutic effect and is it validated?
ii] Useful Pre-clinical Pharmacokinetics Data:-
• Pharmacokinetics parameters from several species, especially species used for toxicology studies.
• Probable site of absorption, and evidence of suitable absorption.
• Routes of elimination, evidence of first-pass metabolism, nonlinear pharmacokinetics, and enzyme induction or inhibition.
• Active/toxic metabolites, metabolic paths.
• Probable isoenzymes involved in metabolizing from in vitro P-450 profiling, and uniqueness of metabolizing enzymes between two species.
• Protein binding and evidence of saturable binding.
iii] Useful Preclinical Toxicology and Toxicokinetic Data:
• Maximally tolerated dose and resulting plasma concentration in two species (rodent and nonrodent).
• Dose and plasma concentrations that produced the first untoward effect.
• Mechanism of toxic effect.
• Toxicity profile and organs involved.
iv] Pharmacokinetic Evaluation:
Pharmacokinetic evaluation typically includes noncompartmenal analysis to characterise pharmacokinetics in term of AUG or clearance, Cmax, Tmax, Vd , and half-life .The high doses used in dose tolerance studies provide an unique opportunity to study linearity at concentrations that might only occur through overdosing in the clinical setting. Because of the relatively small number of subjects per dose level and because most designs involve separate groups of subjects receiving each dose, intersubject variability may mask some deviations from linearity.
v] Pharmacodynamic Evaluation, Pharmacokinetic/Pharmacodynamic modeling:
If the desired pharmacological effects are objective, quantitative, and the same as the therapeutic intent (e.g., increasing gastric pH to treat heartburn), then incorporating pharmacodynamic evaluations into Phase I studies can be extremely useful. If therapeutic effects are indirect or subjective (e.g., treatment of schizophrenia), there might not be a pharmacodynamic marker for the desired therapeutic effect, or it may only be meaningful or measurable in patients with the disease of interest. The challenge of Phase I pharmacodynamic is to carefully select the drug effect that best represents the desired therapeutic response, and to develop methods to measure it reproducibly with minimum variance. If this can be done, Phase I studies offer excellent opportunities for pharmacokinetic/pharmacodynamic modeling. This modeling can be used to confirm or validate preclinical pharmacokinetic/pharmacodynamic models over a wide range of doses. Areas of uncertainty in the preclinical model can be explored, and confirmation of whether the dose or concentration-effect relationship is direct or indirect may be established. As with pharmacokinetics, variability in pharmacodynamic should be assessed. Once validated in Phase I, the pharmacokinetic/pharmacodynamic model can be useful in extrapolating beyond existing data to maximize the chance that effective and safe dosage regimens can be introduced as early as possible in Phase II studies.
Phase II(Therapeutic exploratory trials):
The process of testing an investigational drug on a small number of subjects to show it is generally safe and effective in treating or preventing a specific condition or illness.
Scope: A limited number of studies that help establish the drug’s safety and effectiveness and identify its side effects.
· Subjects: Patients who have the condition or illness to be treated.
· Number of volunteers: Several hundred
· Duration: Several months to 2 years
· Goal: To establish how doctors should use the drug to treat patients with the specified condition or illness.
Prerequisites for Phase II: Preclinical prerequisites for Phase II are similar for Phase I, but with an emphasis placed on understanding preclinical models for efficacy. It is not possible to formulate a checklist for preclinical work required to support Phase II. Drugs that are new but have a known mechanism of action that has been validated in humans may require only minimal preclinical work. Drugs exploiting a known mechanism of action but with a new point of attack require more extensive pharmacological testing. New drugs with novel mechanisms of action will require extensive preclinical investigation. With truly innovative drugs based on new approaches, the validation of the preclinical model for efficacy will only come after the pivotal clinical studies. Therefore, it is vital that preclinical models provide pharmacodynamic measures that can also be applied in Phase II studies as markers of therapeutic efficacy.
Study Design:
Phase II studies are often conducted under optimal experimental conditions, using a relatively homogeneous group of uncomplicated patients (no concomitant illness, medication, or organ dysfunction) and doses close to MTD. These studies are closely monitored and tend to be short term. The number of subjects is relatively small, usually involving only one or two hospitals. Phase IIb studies are larger, involving more centers and a more diverse patient population. Phase II studies are typically double blind and, when possible, are "controlled," i.e., involve comparisons to a placebo and/or standard therapy. Elements of study design are similar to phase I, but specifics will depend on the preclinical data and the pharmacokinetic, pharmacodynamic, and safety data obtained in Phase I, as well as the nature of the disease under investigation. If efficacy parameters are quantitative and objective, then one approach is to use a crossover design with the control agent(s) as treatment alternatives, if the disease has large natural interpatient variability (e.g., arthritis) or subjective efficacy parameters (e.g., schizophrenia), then parallel designs with separate patient groups on active, placebo, or comparator drugs are common.
Phase III (Therapeutic confirmatory trials):
The process of testing an investigational drug on a larger population of subjects to show that it is safe and effective in treating or preventing a specific condition or illness.
- Scope:A large number of studies designed to establish a drug’s safety and effectiveness and to identify its side effects.
- Subjects: Patients who have the condition or illness to be treated, who may have other illnesses, and who may be taking medications in addition to the investigational drug.
- Number of volunteers: Several thousand.
· Duration: Several months to several years.
· Goal: To establish how doctors should use the drug to treat patients with the specified condition or illness.
Population Pharmacokinetics:
To characterize the pharmacokinetics and pharmacodynamic variability of a drug adequately, it needs to be studies in a representative population using relatively large numbers of patients. Considering all the volunteers participating in clinical studies, patients in phase III studies are most representative of the populationintended for treatment. Additionally, the patient population in Phase III is generally larger and more heterogeneous than that encountered in Phase I and II, so it becomes possible to examine the effects of various patient characteristics (e.g. age, body size, renal function, concomitant medications). Therefore, the Phase III patient population is an ideal target for pharmacokinetic and pharmacodynamic scrutiny. However, there are many practical reasons intensive plasma sampling, like that employed to characterise pharmacokinetics in Phase I, is not possible in Phase III studies. Sparse sampling, i.e., one to six plasma samples per patient obtained over the entire course of the study, is usually possible. Through the techniques of population pharmacokinetics, particularly nonlinear mixed effects modeling, these data can be used to characterise pharmacokinetics, quantify variability, and examine its sources.
Phase IV (Post marketing trials):
It is the process of testing an approved drug in order to determine additional information.
- Scope: Multiple studies to determine how a drug compares with other drugs, to ascertain its safety in large number of patients and to identify its effects in patients with conditions that were excluded from phase III trials.
- Subjects: Patients who have the condition or illness to be treated, who may have other illnesses, and who may be taking medications in addition to the study drug.
- Number of volunteers: Several thousand.
- Duration: Several months to several years.
- Goal: To determine if the drug’s use can be broadened to include more patients and/or whether additional safety issues need to be addressed.
Phase II and Phase III both assess the safety and effectiveness of the investigational drug and often involve controlled studies in which the study drug is compared to a placebo (sugar pill) or to an existing drug. This comparison helps minimize bias in interpreting the study results. However, this does not mean that those in the placebo group never receive the investigational medication.
Phase III clinical trials are the most significant because they involve a large number of people who receive treatment for an extended period of time. The outcome of phase III trials provides the basis for FDA approval and establishes exactly how the drug is to be used.
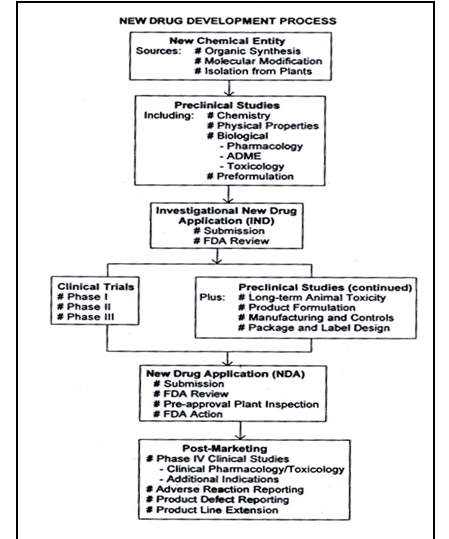
Figure No.1 Schematic representation of new drug development, from drug discovery though clinical studies, NDA and post marketing activities.
Randomized trials2,7:
In a randomized trial, half the patients are chosen at random to get the treatment being tested. They are called the treatment group (or investigational group). In some studies, you will not know which group you are in. These are called blinded studies.
· Single-blindstudy:-In this study, patients do not know whether they are in the treatment group or the control group.
· Double-blindstudy:-In this study, neither the patients nor their doctors know which group they are in.
The purpose of blinded studies is to make sure the results are not biased by anyone's hopes for a certain treatment. Whichever group you are in, you will get the best care possible.
Clinical trials are designed to answer a specific question about a treatment, usuallythe safety and efficacy (how well it works under optimal conditions) of the treatment. Volunteers who meet specific criteria, including having the condition being studied, receive an explanation and, if they choose, join the trial. This informed consentprocess should include explaining the random treatment assignment as well as the risks and possible benefits of the trial and often includes a written form to document those issues and the volunteer’s consent. In a double-blind trial, neither the clinicians caring for the patients nor the participating volunteers know who has been assigned to the active treatment until the trial is concluded (unless a medical problem requires that the information be released before that). In a single-blindtrial, the investigators know the treatment assignments but the participants do not. Blinding procedures protect against the influence of bias(prejudice) for or against the treatment being studied.
Placebos1,8:
A placebo is an inactive treatment designed to resemble the treatment being studied. An example of a placebo is a pill containing sugar instead of the drug being studied. By giving one group of participants a placebo and the other group the active treatment, the researchers can compare how the two groups respond. This gives the researchers a true picture of the active treatment's effects.
Another type of placebo, called a "sham," is used when the treatment under study is a procedure (e.g., acupuncture), not a drug or other substance. A sham procedure is designed to simulate the active treatment but does not have any active treatment qualities. For example, in a clinical trial of acupuncture, the sham procedure might consist of placing acupuncture needles in areas of the body that are not expected to have any therapeutic response.
Placebos are necessary because many factors other than the treatment being studied can influence either the course of an illness or the response of a patient to treatment. For example, many illnesses or symptoms resolve on their own, and interactions with the provider or a patient's expectations about the treatment may influence the patient's response. These and other factors are part of what is known as the "placebo effect," which researchers try to separate from the effects of the treatment they are studying.
Clinical Trial Monitoring 5:
a) Trials are monitored using appropriately trained and qualified individuals. The sponsor will have procedures for these individuals to report on the progress of the trial including possible scientific misconduct.
b) These individuals verify compliance with good clinical practices, including (but not limited to) adherence to the clinical trial protocol, enrollment of appropriate research participants, and the accuracy and complete reporting of clinical trial data.
c) If a sponsor learns that a clinical investigator is significantly deficient in any area, it will either work with the investigator to obtain compliance or discontinue the investigator’s participation in the study, or notify the relevant authorities as required.
Inclusion/Exclusion Criteria5:
All clinical trials have guidelines about who can participate. These are specified in the inclusion/exclusion criteria. Factors that allow someone to participate in a clinical trial are "inclusion criteria." Those that exclude or not allow participation are "exclusion criteria." These criteria are based on factors such as age, gender, the type and stage of a disease, previous treatment history, and other medical conditions. Before joining a clinical trial, a participant must qualify for the study. Some research studies seek participants with illnesses or conditions to be studied in the clinical trial, while others need healthy volunteers. Some studies need both types. Inclusion and exclusion criteria are not used to reject people personally; rather, the criteria are used to identify appropriate participants and keep them safe and to help ensure that researchers can answer the questions they want answered.
Process after the Clinical Trial:1
The researchers carefully analyse the data from the trial and then consider what their findings mean. If the trial has been completed and the results have medical importance, the researchers share their findings with the medical community and the public. The results are usually reported in a peer-reviewed medical journal ("peer-reviewed" means that the report is reviewed before publication by a group of experts in the same field) and/or discussed at scientific meetings. The media may also cover the results of the study. The research team also will inform the participants about the study results soon after the study is completed and all its data are analyzed. Participants should ask the study team when they expect to know the results. A treatment that has been found to be safe and effective in a carefully conducted clinical trial may become a new standard practice.
Safety of participants protected:
There are some risks to participants in clinical trials, but the federal government has imposed mandatory safeguards to protect them. Each clinical trial is reviewed by an Institutional Review Board (IRB), a diverse group of people that must approve the trial. The IRB periodically reviews the clinical trial operations to ensure that the risks are as low as possible, and worth the potential benefits. Some trials also have community advisory boards. In addition, all study participants must read and sign informed consent documents. These documents ensure that participants understand the risks and potential benefits as well as their rights and responsibilities should they decide to participate in the study.
Potential problems in clinical trials:
- Firstly there is no common acceptance from experts upon the definition of the clinical research.
- Secondly, an imperfect public understanding of clinical research, it means that a relatively small percentage of people who volunteer to participate in clinical research trials for example, for adult patients with cancer, the volunteer rates are only five percent.
- Third problem is that the data are inadequate to tell whether investment in clinical research is being well spent or not.
- Fourth problem is about the insufficient funding in certain areas of clinical trials.
- Fifth and sixth problem is related to work force issues involved in clinical trial.
- The seventh problem is poor coordination between Health Management Organizations (HMOs) and the academic medical centers, between schools of nursing and schools of medicine on the other.
Payment to Research Participants:
- Research participants provide a valuable service to society. They take time out of their daily lives and sometimes incur expenses associated with their participation in clinical trials. When payments are made to research participants.
- Any proposed payment should be reviewed and approved by an independent IRB/EC.
- Payments should be based on research participants, time and/or reimbursement for reasonable expenses incurred during their participation in a clinical trial, such as parking, travel, and lodging expenses.
- The nature and amount of compensation or any other benefit should be consistent with the principle of voluntary informed consent.
Payment to Clinical Investigators:
Payment to clinical investigators or their institutions should be reasonable and based on work performed by the investigator and the investigator’s staff, not on any other considerations.
i. A written contract or budgetary agreement should be in place, specifying the nature of the research services to be provided and the basis for payment for those services.
ii. Payments or compensation of any sort should not be tied to the outcome of clinical trials.
iii. Clinical investigators or their immediate family should not have a direct ownership interest in the specific pharmaceutical product being studied.
iv. Clinical investigators and institutions should not be compensated in company stock or stock options for work performed on individual clinical trials.
v. When enrollment is particularly challenging, reasonable additional payments may be made to compensate the clinical investigator or institution for time and effort spent on extra recruiting efforts to enroll appropriate research participants.
vi. When clinical investigators and their staff are required.
vii. To travel to meetings in conjunction with a clinical trial, they may be compensated for their time and offered reimbursement for reasonable travel, lodging, and meal expenses. The venue and circumstances should be appropriate for the purpose of the meeting.
CONCLUSION:
In conclusion, we have came to know about ethical data behind Clinical trials, institution review board/independent ethics committee, ICH GCP, role of FDA, investigator and institution,documents required for the conduct of a clinical trial, Clinical trial Protocol, types of clinical trials, phases of clinical trials, experimental methodology. Clinical trials are designed to answer a specific question about a treatment, usually the safety and efficacy (how well it works under optimal conditions) of the treatment. Volunteers who meet specific criteria, including having the condition being studied, receive an explanation and, if they choose, join the trial. This informed consent process should include explaining the random treatment assignment as well as the risks and possible benefits of the trial and often includes a written form to document those issues and the volunteer’s consent. There are many regulatory requirements for new drug development and approval. Clinical trials are an integral part of the new drug discovery and development process. Before a new medicine can be made available, evidence of its safety and effectiveness must be provided by well-designed, well-controlled, and carefully monitored clinical studies in healthy volunteers and/or patients consenting to participate. Beyond conducting high-quality trials, Roche is committed to providing healthcare stakeholders with full transparency on risks associated with its clinical trials and ensuring the protection of patient safety as well as patients’ personal data. In all cases, the procedures and results mustbe documented appropriately. From a regulatory perspective, if the research is notdocumented, for all intents and purposes, it has not been done.At the completion of the clinical trials conducted using an investigational new drug,and the completion of all nonclinical studies being conducted contemporaneously,New Drug Application (NDA) is filed. The regulations pertaining to NDAs are located in 21 CFR 314 and, as for INDs,provide detailed guidance for both content and format.Typically, sponsors meet with the FDA to discuss the content and format ofan NDA prior to its preparation.
REFERENCES:
1. wikipedia.org
2. clinicalstudyresults.org
3. cdsco.in
4. ichgcp.net
5. Guideline for Good Clinical Practice, (2002), European Medicines Agency, ICH Topic E 6 (R1), 5-48
6. Turner JR., (2007), New drug development: Design, Methodolog and analysis, John Wiley & Sons, Inc., New Jeney, 41-201.
7. Torpy JM., Lynm C., (2006), The Journal of the American Medical Association (JAMA), 295( 23), 2810-2812
8. clinicaltrials.gov
9. Chow SC, Liu JP. (1998), Design and Analysis of Clinical Trials: Concepts and Methodologies, John Wiley and Sons; New York
10. ask.com








