
 ABOUT AUTHORS:
ABOUT AUTHORS:
Bhatt Anjali*, Goshwami Lakshmi, Papola Vibhooti, Pathak Namita, Rawat Shuveksha
Shri Guru Ram Rai Institute of Technology and Science,
Dehradun 248001, Uttrakhand India
*anjali23mar@gmail.com
ABSTRACT
Cyclodextrins were first described by Villiers in 1891. Schardinger laid the foundation of the cyclodextrin chemistry in 1903 and identified cyclodextrin. In the 1930s, Freudenberg identified cyclodextrin and suggested that larger cyclodextrins could exist. Freudenberg and co-workers showed that cyclodextrins were cyclic oligosaccharides formed by glucose units and somewhat later Cramer and co-workers described their ability to form inclusion complexes. By the early 1950s the basic physicochemical characteristics of cyclodextrins had been discovered, including their ability to solubilize and stabilize drugs. The first cyclodextrin-related patent was issued in 1953 to Freudenberg, Cramer and Plieninger. However, pure cyclodextrins that were suitable for pharmaceutical applications did not come available until about 25 years later and at the same time the first cyclodextrin-containing pharmaceutical product was marketed in Japan. Later cyclodextrin-containing products appeared on the European market and in 1997 also in the US. This review aims to assess the use of cyclodextrin in newer drug delivery system such as nanosponges, nanoparticles, nanospheres, liposomes and other drug delivery system.
Reference Id: PHARMATUTOR-ART-1980
INTRODUCTION
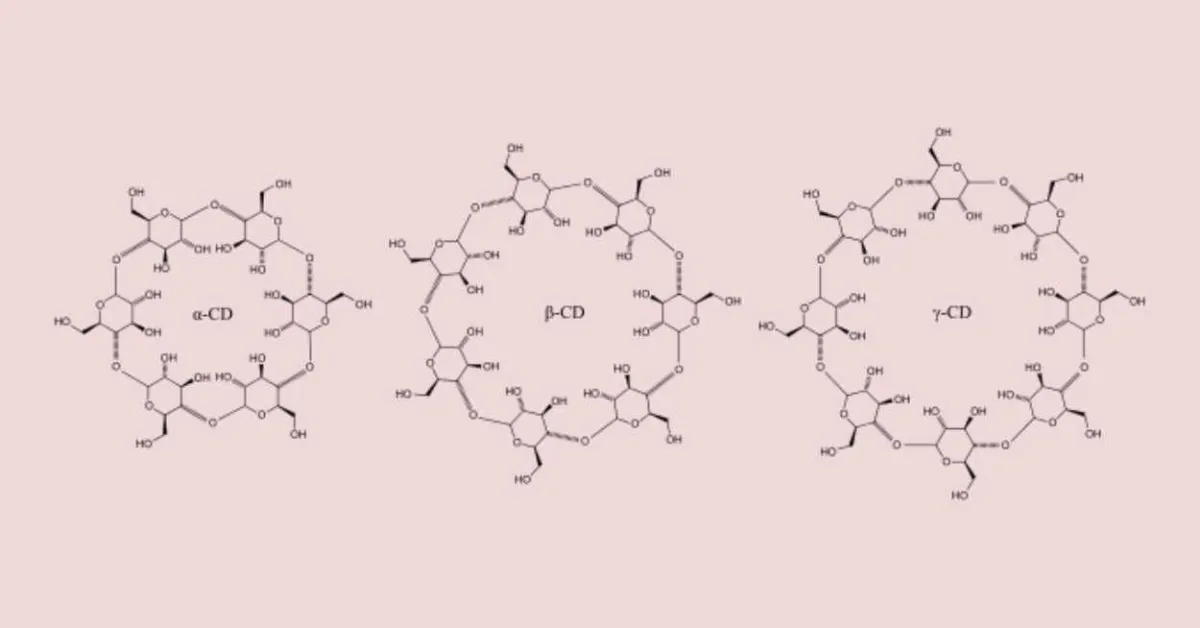
Carbohydrates, such as cellulose, starch and sucrose, are probably the most abundant organic substances in nature and form very ancient time they have been used for shelter, clothing and food. For thousands of years humans have processed carbohydrates through fermentation and observed their enzymatic degradation 1. It is now known that these processes lead to formation of mixtures of monosaccharides, disaccharides and various oligosaccharides, such as linear and branched dextrins and that, under certain conditions, small amounts of cyclic dextrins or cyclodextrins are also being formed during these degradation processes. Cyclodextrins are cyclic (R-1,4)-linked oligosaccharides of R-D-glucopyranose containing a relatively hydrophobic central cavity and hydrophilic outer surface. Owing to lack of free rotation about the bonds connecting the glucopyranose units, the cyclodextrins are not perfectly cylindrical molecules but are toroidal or cone shaped. Based on this architecture, the primary hydroxyl groups are located on the narrow side of the torus while the secondary hydroxyl groups are located on the wider edge. The most common cyclodextrins are R-cyclodextrin, â-cyclodextrin, and ç-cyclodextrin, which consist of six, seven, and eight glucopyranose units, respectively.2
APPROACHES FOR MAKING INCLUSION COMPLEX 3,4,5
The methods generally preferred are:-
I. Kneading
The method involves the formation of paste of cyclodextrin with guest molecules by using small quantity of either water or ethanol to form kneaded mass. Kneaded mass can be dried at 45°C and pulverized.
II. Melting
Excess quantity of guest melted, mixed with powdered cyclodextrin, after cooling excess quantity of quest is removed by washing with weak complex forming solvent. The method restricted to sublimable guest like menthol.
III. Solution-enhanced dispersion by the Supercritical fluids (SEDS)
SEDS is novel, single step method, which can produce solid drug-cyclodextrin complexes. The optimization of processing conditions is essential in order to achieve the optimum complexation efficiency and to compare with drug-cyclodextrin complexation methods described earlier in the literature (e.g. kneading, freeze drying, spray drying etc). Advantages over other methods are (a) Preparation of solid-cyclodextrin complexes in single step process, (b) Achievement of high complexation efficiency (avoidance of excess cyclodextrin in powder). (c) Possibility to minimize the contact of drug with cyclodextrin during the process. (d) Achievement of enhanced dissolution rate of the drug (which is comparable to the dissolution behavior of micronized drug-cyclodextrin complex).depends on the temperature of guest and it must be optimized for every guest.
IV.Co-evaporation / Solvent evaporation method
To the alcoholic solution of guest, aqueous solution of host is added and stirred for sometimes and evaporated at room temp until dried mass obtained, pulverized and sieved and fraction is collected.
V. Microwave Irradiation
This method is developed for rapid organic synthesis and reactions, which require shorter reaction time and higher aim product.
VI. Freeze Drying / Lyophilisation technique
The required stoichiometric quantity of host and guest were added to aqueous solution of cyclodextrin and this suspension stirred magnetically for 24 hours, and resulting mixture is freeze dried at 60°C for 24 hours.
VII. Spray drying / Atomisation
In this method, host solution prepared generally in ethanol: water 50% v/v. To this guest is added and resulting mixture is stirred for 24 hr. at room temperature solution is spray dried by observing following conditions-air flow rate, atomizing air pressure, inlet temperature, outlet temperature, flow rate of solution etc. Product obtained by passing through 63-160 micrometer granulometric sieve.
CD APPLICATION IN DRUG DELIVERY
Oral Drug Delivery
Applications of CDs in oral drug delivery include improvement of drug bioavailability due to increased drug solubility, improvement of rate and extent of dissolution, and/ or stability of the drug at the absorption site, eg, the gastrointestinal tract (GIT) or in formulation, reduction of drug induced irritation, and taste masking). CDs enhance the mucosal drug permeability mainly by increasing the free drug availability at the absorptive surface. CD complexation can provide better and uniform absorption of low-soluble drugs with poor and erratic absorption and also enhance the drug activity on oral administration. CD complexation increased the anthelmentic activity of albendazole and provided a high plasma concentration of the active metabolite. CD complexation increased the absorption of poorly water-solubledrugs, delivered via buccal or sublingual mucosa. Complexation of miconazole, econazole, and clotrimazole with HP-β-CD and genuine CDs increased the toxicity of these drugs on a human buccal cell culture model (TR146) by causing drug supersaturation. Captisol or (SBE) 7m-beta-CD, a solubilizer with osmotic property, was used to design osmotic pump tablets of chlorpromazine and prednisolone. Complexation can also mask the undesirable taste of drugs. Complexation with CDs suppressed the bitter taste of oxyphenonium bromide. HP-β-CDs were shown to have a better oral safety profile than β-CD and other parent CDs, but only limited data are available on the oral safety of the methylated CDs. β-CD is the most cost-effective compound of all CDs, whereas HP-β- and SBE-β-CDs are more expensive( table no 1). 6
Parenteral Drug Delivery
CD derivatives such as amorphous HP-β- and SBE-β-CDs have been widely investigated for parenteral use on account of their high aqueous solubility and minimal toxicity. The solubilizing potentials of both SBE-β- and HP-β-CDs for the drugs melphalan and carmustine were qualitatively similar but the intrinsic reactivities were significantly less with SBE-β-CD.Applications of CDs in parenteral delivery are solubilization of drugs, reduction of drug irritation at the site of administration, and stabilization of drugsunstable in the aqueous environment. Singla et al discussed the use of CDs to enhance the solubility and stability of paclitaxel in formulations and mentioned that the approach needs further research to overcome the serious limitations of CD-based formulations.An IM dosage form of ziprasidone mesylate with targeted concentration of 20 to 40 mg/mL was developed by inclusion complexation of the drug with SBE-β-CD.7 Aqueous phenytoin parenteral formulations containing HP-β-CD exhibited reduced drug tissue irritation and precipitating tendency because their pH values were significantly closer to the physiological value (7.4). Effects of CDs on drug pharmacokinetics were discussed by Rajewski and Stella.8 The synergistic effect of CDs with acids like lactic acid was used to solubilize miconazole for safe parenteral delivery. In some cases complexation may affect drug pharmacokinetics, eg, complexation with sugar branched β-CDs altered the disposition of dexamethsone in mice. The binding values of diflunisal in plasma solutions containing HP-β-CD were found to be lower than the theoretical because of competitive displacement of the drug from the CD by plasma cholesterol. In rabbits, coadministration of M-β-CD with doxorubicin resulted in reduced distribution half-life and modified renal and hepatic distribution profiles of the drug, but the main pharmacokinetic parameters of the CD were unaltered.
Ocular Delivery
Applications of CDs in aqueous eye drop preparations include solubilization and chemical stabilization of drugs, reduction of ocular drug irritation, and enhancement of ocular drug permeability (table no 2). Vehicles used in ophthalmic preparations should be nonirritating to the ocular surface to prevent fast washout of the instilled drug by reflex tearing and blinking. Hydrophilic CDs, especially 2HP-β- and SBE-β- CDs, are shown to be nontoxic to the eye and are welltolerated in aqueous eye drop formulations, eg, increased ocular absorption and shelf life of pilocarpine in eye drop solutions by SBE-β-CD and decreased ocular irritation of a lipophilic pilocarpine prodrug by SBE-β- and HP-β-CDs. The cytotoxicity order of CDs on the human corneal cell line was found to be α-CD 9 DM-β-CD 9 SBE-β-CD = HP-β-CD 9 γ-CD. It was suggested that ocular toxicity with SBE-β-CD (100 mM) after 1 hour of its exposure could be possibly a result of its high osmotic pressure. However, the toxicity with negatively charged SBE-β-CD was greater than that with the control, a neutral hypertonic mannitol solution.CDs enhance drug permeability by making the drug available at the ocular surface. HP-β-CD enhanced the ocular permeability of dexamethasone acetate and also inhibited the conversion of acetate salt to less permeable dexamethasone9. Since only the free drug can permeate biological membranes, ophthalmic delivery of drugs can be limited by the dissociation of drug/CD complexes in the precorneal area due to the limited dilution in this area. The dissociation of drug/CD complexes depends more on the binding of drugs to precorneal proteins, absorption by corneal tissue, and displacement of drugs from CD complexes by precorneal fluid components. Formulation with HP-β-CD, with and without HPMC, improved the bioavailability and maximal mydriatic response of tropicamide by enhancing the drug’s ocular permeability, but reduced the ocular drug irritation probably by maintaining the pH in physiologic range.10 HP-β-CD also enhanced the permeability and miotic response of pilocarpine nitrate without damaging the rabbit corneal tissue.
Nasal Drug Delivery
CDs are effective excipients in nasal drug delivery. CDs improve nasal drug absorption either by increasing aqueous drug solubility and/or by enhancing nasal drug permeability. However, large interspecies differences were found in CD-enhanced nasal drug absorption. DM-β-CD improved the nasal bioavailability of estradiol in rats and rabbits. Nasal absorption of melatonin, a drug with high first pass metabolism was rapid and efficient when administered with β-CD and the peak levels were ~50 times higher than those observed after oral administration. Midazolam was absorbed rapidly when administered as an aqueous nasal spray (pH 4.3) containing SBE-β-CD (14% wt/vol), HPMC (0.1% wt/vol), and other additives. CDs can also be used to reduce the nasal toxicity of other enhancers without affecting their absorption-enhancing property. Salbutamol release from the powder inhaler formulations containing γ-CD and DM-β-CD was faster than that from control with lactose; at the amount studied γ-CD was safer than DM-β-CD. 11
Rectal Drug Delivery
Applications of CDs in rectal delivery include enhancing drug absorption from a suppository base either by enhancing drug release from the base or by increasing drug mucosal permeability, increasing drug stability in the base or at the absorption site, providing sustained drug release, and alleviating drug-induced irritation. The effect of CDs on rectal drug absorption can be influenced by partition coefficient of the drug and its CD complex, magnitude of the complex stability constant, and nature of the suppository base (oleaginous or hydrophilic). Hydrophilic CDs (especially methylated and hydroxylpropyl CDs) enhance the absorption of lipophilic drugs by improving the drug release from oleaginous vehicles and/or by increasing the drug dissolution rate in rectal fluids. Formation of hydrophilic CD complexes was found to inhibit the reverse diffusion of drugs into oleaginous vehicles by reducing the drug/vehicle interaction. Rectal absorptions of flurbiprofen and biphenylacetic acid were improved by DM-β-CD and HP-β-CD, respectively. In the presence of hydroxyl propyl methyl cellulose (HPMC), β-CD markedly reduced the bioavailability of acetaminophen from both aqueous solution and hydrogels by forming a complex with a lower partition coefficient or higher hydrophilicity. CDs enhance the rectal absorption of inabsorbable, hydrophilic drugs such as antibiotics, peptides, and proteins bytheir direct action on the rectal epithelial cells. α-CD enhanced the rectal absorption of morphine and human chorionic gonadotropin by increasing their mucosal permeability and reducing their degradation.12
Controlled Drug Delivery
CDs, due to their ability either to complex drugs or to act as functional carrier materials in pharmaceutical formulations, can serve as potential candidates for efficient and precise delivery of required amounts of drugs to targeted site for a necessary period of time. β-CD derivatives are classified as hydrophilic, hydrophobic, and ionizable derivatives. The hydrophilic derivatives improve the aqueous solubility and dissolution rate of poorly soluble drugs, while the hydrophobic derivatives retard the dissolution rate of water-soluble drugs from vehicles. The ionizable CD derivatives, on the other hand, improve inclusion capacity, modify drug dissolution rate, and alleviate drug irritation, eg, use of CME-β-CD to obtain delayed release– type formulations (table no 3). Highly hydrophilic derivatives, such as 2HP-β-, G2-β-, and SBE-β-CDs were used in immediate release formulations that dissolve readily in the GIT and enhance the oral bioavailability of poorly soluble drugs. Hydrophobic CDs, such as alkylated and acylated derivatives are useful as slow-release carriers in prolonged release formulations of water-soluble drugs. Combination of short-chain and long-chain peracylated β-CDs in an appropriate molar ratio was suggested to be useful to provide an effectively controlled release rate of water-soluble drugs, eg, markedly retarded release rate of molsidomine on complexation with peracylated β-CDs. CDs can also be used along with other carrier materials to optimize drug release rate. Improved nifedipine bioavailability with reduced first pass metabolism was observed from a modified oral dosage form containing a fast releaseportion of the drug with HP–β-CD and HCO-60, a nonionic surfactant (ie, amorphous drug form obtained by spray drying with the CD and surfactant) and a slow release portion with hydroxy propyl celluloses (HPCs) of different viscosity grades.SBE-β-CD has been used in the design of sustained release matrix tablets of poorly soluble drugs. Use of CDs with a hydroxyapatite matrix was indicated to control the release of chemotherapeutic agents containing toxic metals, such as Rhodium II citrate and butyrate, and to provide localized antitumor chemotherapy with minimal side effects.13
Colon-Specific Drug Delivery
CDs are barely hydrolyzed and only slightly absorbed in the stomach and small intestine but are absorbed in the large intestine after fermentation into small saccharides by colonic microbial flora. The peculiar hydrolyzing property of CDs makes them useful for colon drug targeting. Biphenyl acetic acid (BPAA) prodrugs for colon-specific delivery were developed by conjugation of the drug onto one of the primary hydroxyl groups of α-, β-, and γ-CDs through an ester or amide linkage. Drug conjugation with α-CD resulted in a delayed release–type prodrug formulation for colon-specific delivery that alleviates the side effects of drugs while maintaining their therapeutic effect, eg, site-specific degradation of prednisolone/α-CD conjugates in the large intestine alleviated the side effects of the drug while maintaining its anti-inflammatory action.Complexation of triamcinolone acetonide (TA) with β-CD improved the sphericity of microcrystalline cellulose (MCC)–β-CD-TA spherical pellets (5:90:5) prepared by extrusion and spheronization for colon targeting. TA complexation with the CD also facilitated the application of coating resistant to gastric and small intestinal media and maintained the pellet integrity in dissolution medium with no premature bursting of coatings on granule swelling.14
Peptide and Protein Delivery
Various problems associated in practical use of therapeutic peptides and proteins are their chemical and enzymatic instability, poor absorption through biological membranes, rapid plasma clearance, peculiar dose response curves, and immunogenicity.CDs, because of their bioadaptability in pharmaceutical use and ability to interact with cellular membranes, can act as potential carriers for the delivery of proteins, peptides, and oligonucleotide drugs. The existence of efflux pumps may serve as an additional barrier for nonspecific uptake of peptides and thus can cause low peptide bioavailability. The various established mechanisms for CD-improved nasal absorption of peptides are interaction with membrane lipids and proteins in the nasal epithelium that reduces the membrane barrier function, inhibition of proteolytic enzyme activities in the nasal mucosa, and finally inhibition of protein or peptide aggregation by direct action upon these molecules. Since the absorption-enhancing effects of CDs are reversible, as enhancers they are less toxic than other widely used enhancers, eg, the effect of CDs on the nasal cilliary beat frequency were observed to be mild, reversible, and less toxic. However, substantial interspecies differenceswere observed in the absorption enhancement of peptides from CD solutions. DM-β-CD, the only effective nasal absorption enhancer out of the CDs studied (β-, HP-β-, γ-, and DM-β-CDs), largely improved the nasal absorption of insulin and adreno crotico tropic hormone (ACTH) from solutions in rats (bioavailability ~70% to 100%) but in rabbits and healthy human volunteers, the same CD/insulin solution was found to be ineffective. Improved bioavailability of insulin (up to 13%) was observed on nasal administration of powder formulations containing DM-β-CD compared with the control containing lactose instead of the CD. β-CD improved insulin loading of alginate microspheres prepared by an emulsion-based process.15
Gene and Oligonucleotide Delivery
Gene delivery technologists are now testing CD molecules in the hope of finding an optimal carrier for the delivery of therapeutic nucleic acids, however, the limitations of CDs, such as CD-associated toxicity (eg, DM-β-CD) have to be considered before their clinical use. CDs can improve cellular uptake of oligonucleotides and also delay their degradation by increasing their stability against endonucleases. ON-adamatane conjugates associated with HP-β-CD provided significantly increased cellular uptake of ONs. Neutral and amphipilic as well as cationic CDs have been used for synthesis of novel gene delivery vectors. Neutral CDs like β-, DM-β-, and HP-β-CDs were reported to increase DNA cellular uptake by increasing its permeability. The increased DNA permeability was reported to be a result of interaction of the CDs with membrane components such as cholesterol, but not due to their complexing ability for DNA. Cationic polyamino CDs, because of their polycationic polyanionic interaction with mononucleotides, neutralized the multiple charges on DNA and thus made DNA compact into a particle of suitable size for cellular internalization. Amphiphilic CDs, because of their vesicle-forming potential, offer an additional possibility for polar nucleotides to complex into aqueous vesicle core while allowing hydrophobic agents to complex into individual cavities or interior of the bilayer with multiple lipophilic hydrocarbon chains. Polyplexes (polycation polymer/DNA composite structures) of linear, cationic, β-CD–containing polymers (βCDPs) were found to be suitable for DNA delivery due to their increased transfection efficiency and stability against enzymatic degradation with low in vitro and in vivo toxicity. CDs were also found to enhance plasmid or viral-vector– based delivery of genes. Positively charged quarternary amino and tertiary amino β-CDs significantly enhanced the transfection efficiency of negatively charged adenoviral vector-based gene formulations. It was reported that the transfection enhancement by the cationic β-CDs could be a result of increased viral internalization caused by increased viral binding to cell and improved cell membrane permeability.16,17,18,19
Dermal and Transdermal Delivery
CDs have been used to optimize local and systemic dermal drug delivery. Applications of CDs in transdermal drug delivery include enhancement of drug release and/or permeation, drug stabilization in formulation or at absorptive site, alleviation of drug-induced local irritation, sustaining of drug release from vehicle, and alteration of drug bioconversion in the viable skin. Although the drug partition coefficient (eg, a lipophilic drug) may be decreased on complexation with CDs (eg, with hydrophilic CDs), the increased drug solubility and thermodynamic activity in vehicles can lead to increased drug permeability through skin, eg, increased skin permeability of dexamethasone by HP-β-CD. β- and HP-β-CDs significantly enhanced the anti-inflammatory effects of indomethacin in hydroxyethyl cellulose hydrogels in healthy volunteers. CDs enhance drug delivery by increasing the drug availability at the barrier surface, where the free drug partitions from the CD cavity into lipophilic barrier. The free drug fraction at the barrier surface depends on the drug dissolution rate, relative magnitude of the stability constants of the CD complexes with the drug and the competing agent at the absorption site, and the drug absorption rate constant. For absorption, the CD complex has to dissociate to release free drug, the actual absorbable species and the dissociation of CD complex depends on the magnitude of the complex stability constant. If the complex stability constant is too high, the complex may not release the free drug at the absorptive site and thus may decrease orinhibit drug absorption.The effect of HP-β-CD concentration on the iontophoretic delivery of hydrocortisone (ie, higher drug amount delivered at lower CD concentration [1%] compared with higher CD concentrations [3% or 5%]) indicated the delivery of only the free drug with the CD acting as a carrier. β- and HP-β-CDs increased the skin permeability of dexamethasone and also improved its stability in skin by protecting it against skin metabolism. CDs, by increasing solubility, facilitate drug incorporation into formulation and thus increase the drug concentration in the formulation. HP-β-CD increased the amount of piroxicam transported through skin but pretreatment of skin with the CD showed no effect on drug retention in skin. Hence the CD effect on the drug’s skin permeability was reported to be due to increased drug concentration in gel and not due to enhancement of drug iontophoretic flux. Nitroglycerin complexation with DE-β-CD accelerated the drug release rate from ointments but the same with β-CD retarded the drug release. Hence a combination of the drug complexes with DE-β-CD and β-CD was suggested to obtain sustained release percutaneous preparations of the drug. CDs, the safer solubilizing agents with bioadaptability and multifunctional characteristics, have been evaluated for formulation of poorly water-soluble cosmetic materials. HP-CDs increased the aqueous solubility of cosmetic materials, retarded the release rate of fragrance materials with no toxicity in topical liquid preparations,and also reduced permeation rate of eugenol and methyl paraben through hairless mouse skin. Other CD applications in cosmetics include masking of smell and stench, stabilization of cosmetic materials (eg, loyal jelly and antiplasmin drugs), assisting in preparation of stable emulsion and suspension, inhibition of foaming caused by amphiphilic materials, and powderization of oily materials. The ability of CDs to increase stability (against light and oxygen) and solubility of sparingly water-soluble molecules made them useful in the formulation of cosmetic products.20-24
Brain Drug Delivery or Brain Targetting
The concept of Bodor’s chemical delivery system (CDS) (ie, covalent coupling of drugs to 1-methyl-1, 4-dihydronicotinic acid through an enzymatically labile linkage, which increases drug lipophilicity) was applied for targeting drugs such as steroids, antitumor agents, and calcium channel antagonists to brain. However, presence of the lipophilic moiety makes prodrugs of CDS poorly water-soluble. HP-β- CD, due to its ability to solubilize drugs and also to enhance the chemical stability of dihydronicotinic acid in aqueous solution solved the solubility problems of CDS. Formulation is an important and integral concern in the development of CDS, especially those for brain targeting. Formulation development of CDS is based on the need for appropriate dosage form, stability, solubility, and dissolution characteristics. HP-β-CD contributed to the development and preclinical testing of several CDS by providing a stable and water-soluble dosage form suitable for parenteral administration. Use of CDs in the formulation of CDS can be demonstrated by the significantly improved solubility, stability, and pharmacologic activity of CDS of thyrotropin-releasing hormone analogs on complexation with HP-β-CD.The very low penetration across the BBB greatly hinders the therapeutic use of peptides, and whenever unexplainable poor peptide absorption is seen the role of the efflux pumps should be examined. It was reported that P-gp– mediated peptide transport may play an important role in greatly reducing the peptide delivery to the central nervous system in vivo. It was also indicated that CDs such as DM-β-CD, due to their inhibitory effect on P-gp efflux function, may enhance drug delivery to brain. 25.
SOME NOVEL DRUG DELIVERY SYSTEM
Liposomes
In drug delivery, the concept of entrapping CD-drug complexes into liposomes combines the advantages of both CDs (such as increasing the solubility of drugs) and liposomes (such as targeting of drugs) into a single system and thus circumvents the problems associated with each system. Liposomes entrap hydrophilic drugs in the aqueous phase and hydrophobic drugs in the lipid bilayers and retain drugs en route to their destination. The fact that some lipophilic drugs may interfere with bilayer formation and stability limits the range and amount of valuable drugs that can be associated with liposomes. By forming watersoluble complexes, CDs would allow insoluble drugs to accommodate in the aqueous phase of vesicles and thus potentially increase drug-to-lipid mass ratio levels, enlarge the range of insoluble drugs amenable for encapsulation (ie, membrane-destabilizing agents), allow drug targeting, and reduce drug toxicity. To encapsulate large amounts of lipophilic drugs in liposomes, a CD molecule forming an inclusion complex with a high drug:CD ratio should be selected. Liposomal entrapment of prednisolone was higher when incorporated as HP-β-CD complex than as free drug. Selection of CD can also have a significant effect on the amount of drug associated with vesicles, eg, HP-β-CD, with a more lipophilic interior and considerably higher aqueous solubility incorporated higher drug amounts in vesicles than β-CD. However, HP-β-CD, as a result of its ability to get entrapped in higher amounts in the vesicles, also showed a higher velocity of destabilizing effect on vesicles than β-CD.Complexation with CDs can improve the stability of liposomes, eg, most stable liposomal formulations of metronidazole and verapamil were obtained by direct spray drying of lipid, drug, and HP-β-CD mixture.26-27
Microspheres
In the presence of a high percentage of highly soluble hydrophilic excipients, complexation may not improve the drug dissolution rate from microspheres. Nifedipine release from chitosan microspheres was slowed down on complexation with HP-β-CD in spite of the improved drug-loading efficiency. Since it is highly unlikely for CD molecules to diffuse out of the microspheres, even with a low stability constant, the complex must first release the free drug that can permeate out of the microspheres. Hence the observed slow nifedipine release from the microspheres was reported to be due to lesser drug availability from the complex andalso due to formation of hydrophilic chitosan/CD matrix layer around the lipophilic drug that further decreases the drug matrix permeability. Sustained hydrocortisone release with no enhancement of its dissolution rate was observed from chitosan microspheres containing its HP-β- CD complex. CDs were also used to modulate peptide release rate from microspheres, eg, HP-β-CD coencapsulation in PLGA microspheres slowed down insulin release rate. Microspheres, prepared by spray drying of a water/oil emulsion containing the CD provided constant insulin release up to 45 days without initial burst and maintained the peptide stability during the entire release phase. The slowing down of overall release rate of the peptide was reported to be due to its decreased matrix diffusivity caused by its higher apparent molecular weight and size on complexation. Co-encapsulation of the CD also reduced the apparent particle size of the microspheres. A high entrapment efficiency of gabexate mesylate (GM) was observed with all types of bioadhesive and biodegradable starch/CD microspheres prepared by chemical crosslinking of an alkaline solution of a mixture of starch and CD (α-, β-, or γ-CD) with epichlorohydrin. The amount of GM included and its proportion in microspheres after storage were in agreement with its affinity for the CDs and the order of association constants of its complexes.28-29
Microcapsules
It was suggested that crosslinked β-CD microcapsules, because of their ability to retard the release of water-soluble drugs through semipermeable membranes, can act as release modulators to provide efficiently controlled release of drugs. Terephthaloyl chloride (TC) crosslinked β-CD microcapsules were found to complex p-nitrophenol rapidly and the amount complexed increased as the size of the microcapsules decreased. TC crosslinked β-CD microcapsules retarded the diffusion of propranolol hydrochloride through dialysis membrane. Double microcapsules, prepared by encapsulating methylene blue with different amounts of β-CD microcapsules inside a crosslinked human serum albumin (HSA), showed decreasing release rate of methylene blue with increasing amount of β-CD microcapsules. Dissociation of methylene blue complex with β-CD microcapsules was found to serve as an additional mechanism in controlling the release kinetics of HAS double microcapsules. In the case of HSA microcapsules with parent β-CD, the hydrating property of the CD, by promoting the diffusion of water into the microcapsules, caused an increased release rate of methylene blue compared with those without the CD. However, in the case of HSA double microcapsules (ie, with β-CD microcapsules), the hydrophobic groups introduced during crosslinking suppressed the CD hydration and provided controlled release without enhancing the diffusion of water that can impair the complexation of methylene blue.30
Nanoparticles
Two applications of CDs have been found very promising in the design of nanoparticles: one is increasing the loadingcapacity of nanoparticles and the other is spontaneous formation of either nanocapsules or nanospheres by nanoprecipitation of amphiphilic CDs diesters.CDs increased the loading capacity of poly (isobutylcyanoacrylate) nanoparticles. The increased loading capacity was reported to be a result of increased drug concentration in the polymerization medium on addition of the drug:CD complex and increased number of hydrophobic sites in the nanosphere structure on association of large amounts of CDs to the nanoparticles. HP-β-CD increased saquinavir loading into poly (alkylcyanoacrylate) nanoparticles by providing a soluble drug reservoir in polymerization medium that feeds the nanoparticle-formation process. A dynamic equilibrium was observed between the complex, the dissociated species, and the forming polymeric particle. It was indicated that during nanoparticle formation the free drug gets progressively incorporated into polymer network, driven by the drug partition coefficient between the polymer and polymerization medium though there may be a simultaneous direct entrapment of some drug/CD complex. Addition of HP-β-CD in the polymerization medium of poly (ethylcyanoacrylate) (PECA) nanospheres improved the subcutaneous absorption of metoclopramide in rats. PECA nanospheres with HP-β-CD provided the highest drug concentration and enhanced drug absorption compared with those with dextran or with drug solution. Loading techniques and also the type of β-CDa can influence the loading and release properties of the nanospheres. Inclusion complexation of progesterone with β-CDa prior to its entrapment in nanospheres increased the drug loading into nanospheres. Progesterone-loaded β-CDa nanospheres acted as a promising nonsurfactant injectable delivery system to provide rapidly a high quantity of the water-insoluble drug (within 1 hour).31-34
CONCLUSION
CDs, as a result of their complexation ability and other versatile characteristics, are continuing to have different applications in different areas of drug delivery and pharmaceutical industry. However, it is necessary to find out any possible interaction between these agents and other formulation additives because the interaction can adversely affect the performance of both. It is also important to have knowledge of different factors that can influence complex formation in order to prepare economically drug/CD complexes with desirable properties. Since CDs continue to find several novel applications in drug delivery, we may expect these polymers to solve many problems associated with the delivery of different novel drugs through different delivery routes.
Table 1.Applications of CDs in Oral Delivery
|
Effect |
Drug |
CD |
|
↑Bioavailability by |
β-CD |
Ketoprofen, Griseofulvin, Terfenadine |
|
↑Solubility and dissolution rate |
HP-β-CD SBE7–β-CD DM-β-CD M-β-CD ME-β-CD |
Albendazole, Ketoprofen, Phenytoin, Gliclazide Spiranolactone Tacrolimus Albendazole Phenytoin |
|
↑Intensity or duration of therapeutic activity |
β-CD HP-β-CD |
Terfenadine, Tolbutamide Tolbutamide, Amylobarbitone |
|
↑Permeability |
HP-β-CD |
Flutamide
|
|
↑Gastrointestinal stability |
γ-β-CD HP-β-CD |
Digoxin Rutin |
|
↑Sublingual bioavailability |
HP-β-CD |
Clomipramine, Testosterone |
|
↑Buccal bioavailability |
HP-β-CD SBE7-β-CD |
Danazole |
Table2.Some of the Examples of Usage of CDs in Aqueous Eye Drop Solutions.
|
Acetazolamide |
HP-β-CD, α-CD, HP-β-CD |
|
Arachidonylethanolamide |
HP-β-CD |
|
Cyclosporine |
α-CD |
|
Dexamethasone |
HP-β-CD |
|
Dexamethasone acetate |
HP-β-CD |
|
Diclofenac |
HP-β-CD, M-β-CD |
|
Ethoxyzolamide |
|
|
Pilocarpine |
HP-β-CD, α-CD, β-CD, HP-β-CD, SBE-β-CD |
|
Pilocarpine prodrugs |
HP-β-CD |
Table 3. Applications of Various CD Derivatives in the Formulation of Modified Release Preparations.
|
Derivative |
Drug |
Summary |
|
Diethyl-β-CD |
Diltiazem Buserelin acetate Nitroglycerine Tiaprofenic acid |
Sustained release for oral use Sustained release for subcutaneous use Sustained release for percutaneous use Sustained release Delayed release |
|
Triacetyl-β-CD |
Flufenamic acid |
Prolonged release for oral use |
|
Peracylated-β-CD (TB-β-CD) |
Molsidomine Salbutamol Captopril |
Sustained release for oral use Prolonged release for oral use Sustained release |
|
Al-β-CD |
Sulfate Recombinant human basic fibroblast growth factor |
Sustained release for oral use; enhanced stability |
|
O-carboxymethyl- O-β-CD |
Molsidomine Diltiazem HCl |
Delayed release |
REFERENCES
1. Drug solubilization and stabilization. J Pharm Sci. 1996;85:1017Y1025.
2. Endo T, Nagase H, Ueda H, Kobayashi S, Nagai T. Isolation, purification, and characterization of Cyclomaltodecaose (curly epsilon- Cyclodextrin), Cyclomaltoundecaose (zeta-Cyclodextrin) and Cyclomaltotridecaose (é-Cyclodextrin). Chem Pharm Bull (Tokyo). 1997;45:532Y536.
3. Endo T, Nagase H, Ueda H, Shigihara A, Kobayashi S, Nagai T. Isolation, purification and characterization of Cyclomaltooctadecaose (v-Cyclodextrin), Cyclomaltononadecaose (xi-Cyclodextrin), Cyclomaltoeicosaose (o-Cyclodextrin) and Cyclomaltoheneicosaose (ã-Cyclodextrin. Chem Pharm Bull (Tokyo). 1998;46:1840Y1843.
4. Miyazawa H, Ueda H, Nagase T, Endo T, Kobayashi S, Nagai T. Physicochemical properties and inclusion complex formation of δ- cyclodextrin. Eur J Pharm Sci. 1995;3:153Y162.
5. Szejtli J. Cylodextrin in drug formulations: Part I. Pharm Technol Int. 1991;3:15Y23.
6. Arima H, Yunomae K, Miyake K, Irie T, Hirayama F, Uekama K. Comparative studies of the enhancing effects of cyclodextrins on the solubility and oral bioavailability of tacrolimus in rats. J Pharm Sci. 2001;90:690Y701.
7. Kim Y, Oksanen DA, Massefski W, Blake JF, Duffy EM, Chrunyk B. Inclusion complexation of ziprasidone mesylate with beta-cyclodextrin sulfobutyl ether. J Pharm Sci. 1998;87:1560Y1567.
8. Rajewski RA, Stella VJ. Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. J Pharm Sci. 1996;85:1142Y1168.
9. Cramer F, Saenger W, Satz HC. Inclusion compounds. ΧΙX. The formation of inclusion compounds of alpha cyclodextrin in aqueous solutions, thermodynamics and kinetics. J Am Chem Soc. 1967;89:14Y20.
10. Faucci MT, Mura P. Effect of water-soluble polymers on naproxen complexation with natural and chemically modified beta-cyclodextrins. Drug Dev Ind Pharm. 2001;27:909Y917.
11. Uekama K, Hirayama F, Irie T. Cyclodextrin drug carrier systems. Chem Rev. 1998;98:2045Y2076
12. Matsuda H, Arima H. Cyclodextrins in transdermal and rectal delivery. Adv Drug Deliv Rev. 1999;36:81Y99.
13. Stella VJ, Rajeswski RA. Cyclodextrins: their future in drug formulation and delivery. Pharm Res. 1997;14:556Y56.
14. Irie T, Uekama K. Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. J Pharm Sci. 1997;86:147Y162.
15. Szejtli J. Medicinal applications of cyclodextrins. Med Res Rev. 1994;14:353Y386
16. Redenti E, Pietra C, Gerlozy A, Szente L. Cyclodextrins in oligonucleotide delivery. Adv Drug Deliv Rev. 2001;53:235Y244.
17. Hwang SJ, Bellocq NC, Davis ME. Effects of structure of β-cyclodextrin-containing polymers on gene delivery. Bioconjugate Chem. 2001;12:280Y290.
18. Pun SH, Davis DE. Development of a nonviral gene delivery vehicle for systemic application. Bioconjugate Chem. 2002;13:630Y639.
19. Croyle MA, Roessler BJ, Hsu CP, Sun R, Amidon GL. Beta cyclodextrins enhance adenoviral-mediated gene delivery to the intestine. Pharm Res. 1998;15:1348Y1355.
20. Lopez RF, Collett JH, Bently MV. Influence of cyclodextrin complexation on the in vitro permeation and skin metabolism of dexamethasone. Int J Pharm. 2000;200:127Y132.
21. Orienti I, Zecchi V, Bernabei S, Sentimenti S, Fini A. Diffusion of ketoprofen from coprecipitates through a non porous lipidic membrane.Boll Chim Farm. 1989;128:336Y343.
22. Loftsson T, Masson M. Cyclodextrins in topical drug formulations: theory and practice. Int J Pharm. 2001;225:15Y30.
23. Chang SL, Banga AK. Transdermal iontophoretic delivery of hydrocortisone from cyclodextrin solutions. J Pharm Pharmacol. 1998;50:635Y640.
24. Doliwa A, Santoyo S, Ygartua P. Transdermal iontophoresis and skin retention of piroxicam from gels containing piroxicam: hydroxypropyl- beta-cyclodextrin complexes. Drug Dev Ind Pharm.2001;27:751Y758.
25. Mosher G, Thompson DO. Complexation and Cyclodextrins. In: Swarbrick J, Boylan JC, eds. Encyclopedia of Pharmaceutical Technology. 2nd ed. New York, NY: Marcell Dekker; 2002:531Y558.
26. McCormack B, Gregoriadis G. Drugs-in-cyclodextrins-in-liposomes: an approach to controlling the fate of water insoluble drugs in vivo. Int J Pharm. 1998;162:59Y69.
27. McCormack B, Gregoriadis G. Drugs-in-cyclodextrins-in liposomes: a novel concept in drug delivery. Int J Pharm.1994;112:249Y258.
28. Filipovic-Grcic J, Laan MB, Skalko N, Jalsenjak I. Chitosa microspheres of nifedipine and nifedipine-cyclodextrin inclusion complexes. Int J Pharm. 1996;135:183Y190.
29. Filipovic-Grcic J, Voinovich D, Moneghini M, Becirevic-Lacan M Magarotto L, JalsenjakI. Chitosan microspheres with hydrocortisone and hydrocortisone–hydroxypropyl-b-cyclodextrin inclusion complex. Eur J Pharm Sci. 2000;9:373Y379.
30. Memisoglu E, Bochot A, Sen M, Charon D, Duchene D, Hincal AA. Amphiphilic beta- cyclodextrins modified on the primary face: synthesis, characterization, and evaluation of their potential as novel excipients in the preparation of nanocapsules. J Pharm Sci.2002;91:1214Y1224.
31. Duchene D, Ponchel G, Wouessidjewe D. Cyclodextrins in targeting. Application to nanoparticles. Adv Drug Del Rev. 1999;36:29Y40.
32. Monza da Silveira A, Ponchel G, Puisieux F, Duchene D. Combined poly (isobutylcyanoacrylate) and cyclodextrins nanoparticles for enhancing the encapsulation of lipophilic drugs. Pharm Res. 1998;15:1051Y1055.
33. Duchene D, Ponchel G, Wouessidjewe D. Cyclodextrins in targeting Application to nanoparticles. Adv Drug Deliv Rev.1999;36:29Y40.
34. Boudad H, Legrand P, Lebas G, Cheron M, Duchene D, Ponchel G.Combined hydroxypropyl-beta-cyclodextrin and poly (alkylcyanoacrylate) nanoparticles intended for oral administration of saquinavir. Int J Pharm. 2001;218:113Y124.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








