
 About Authors:
About Authors:
Ravindra K. Kamble, Priyadarshani R. Kamble*
Department of Pharmaceutics and Quality Assurance,
Bhupal Nobles’ College of Pharmacy, Udaipur, Rajasthan, INDIA.
*priya.s.bansode@gmail.com
ABSTRACT
Recent studies reported approval of lipid vesicles as drug carriers for chemotherapeutic agents and bio-actives which have been revealed in a number of lipid vesicles based formulations, which are commercially available or are currently undergoing clinical trials. This review is mainly focused on effectiveness and permeation enhancing controversy of lipid vesicles as dermal and transdermal drug delivery with special emphasis on recent advances in this field, including the development of deformable vesicles, ethosomes and invasomes. Only specially designed lipid vesicles have been shown to be capable of achieving enhanced delivery. The incorporation of additives, such as anionic surfactants and ethanol, fluidize the phospholipid bilayers, thus can penetrate the intercellular pathways of the skin.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1589
INTRODUCTION:
During the past decades there has been wide interest in exploring innovative techniques to increase drug absorption through skin [1, 2]. The skin covers a total surface area of approximately 1.8 m2 and provides the contact between the human body and its external environment. Transdermal drug delivery uses the skin as an alternative route for the delivery of local and systemically acting drugs. This drug delivery route have several advantages compared to oral drug administration. The variables that could influence gastro-intestinal absorption such as pH, food intake, gastro-intestinal motility, the hepatic metabolism leads to the variations in drug plasma levels. Drugs particularly of a narrow therapeutic window can be delivered through skin with reduced the side effects. The barrier nature of skin makes it difficult for most drugs to penetrate and permeate through it [3-5]. The earliest report of liposomes effectiveness for skin delivery was published by Mezei and Gulasekharam(1980), conflicting results continued to be published concerning their effectiveness, enhancing the controversy of liposomes as dermal and transdermal drug delivery vehicles [6]. Classic lipid vesicular systems are of little value as carriers for drug delivery via the skin because they do not penetrate it deeply, but rather remain confined to upper layers of the stratum corneum. Only specially designed liposomes have been shown to be capable of achieving enhanced delivery. The incorporation of additives, such as anionic surfactants and ethanol, can fluidize the phospholipid bilayers, thus increasing the depths to which liposomes can penetrate the intercellular pathways of the skin. Also, liposomes that have been conjugated with PEG or antibodies can increase the residence time of anticancer drugs in the circulation and enhance drug accumulation in tumors. A new class of lipid vesicles is the highly deformable (elastic or ultra flexible) liposomes, which have been termed Transfersomes. Recent studies have reported that deformable liposomes were able to improve in vitro skin delivery of various drugs. Ethosome is another novel lipid carrier, recently developed by Touitou et al., showing enhanced skin delivery [7-10]. The aim of present article is to provide an overview of modified lipid vesicles as carriers for skin delivery of drugs, with special emphasis on recent advances, including the development of deformable liposomes, ethosomes and invasomes.
Conventional lipid vesicles as carriers:
Liposomes have been extensively investigated as potential drug delivery systems due to the enormous diversity of structure and composition that can be achieved [11]. Liposomes are broadly defined as lipid bilayers surrounding an aqueous space (Fig.1). Multilamellar vesicles consist of several (up to 14) lipid layers (in an onion-like arrangement) separated from one another by a layer of aqueous solution. These vesicles are over several hundred nanometers in diameter. Small unilamellar vesicles are surrounded by a single lipid layer and are 25–50 nm (according to some authors up to 100 nm) in diameter. Large unilamellar vesicles are, in fact, a very heterogeneous group of vesicles that, like the Small Unilamellar Vesicles, are surrounded by a single lipid layer. The diameter of these liposomes is very broad, from 100 nm up to cell size (giant vesicles) [12]. The potential value of liposomes for topical therapy was first introduced by Mezei and Gulasekharam (1980). In this study, greater four- to five-fold triamcinolone acetonide concentrations in the epidermis and dermis, with lower systemic drug levels, were observed when the drug was delivered from liposomal lotion in comparison with conventional formulations of the same drug concentration. An important role of the transappendageal route in improving skin delivery of drugs by liposomes was also suggested. Recent studies suggested that the role of the transappendageal route is limited to improved vesicular penetration into (i.e. targeting) but not necessarily through hair follicles and that it plays no major role in improving liposomal transdermal delivery. Furthermore, the composition of the vesicles influences their physico-chemical characteristics such as, size, charge, thermodynamic phase, lamellarity and bilayer elasticity. These physico-chemical characteristics have a significant effect on the behavior of the vesicles and hence on their effectiveness as a drug delivery system. An enhancement of liposomal entrapment and simultaneously a decrease of vesicle diameter were observed [13-16]. Several in vivo and in vitro transport studies reported that conventional liposomes only enhanced skin deposition, with mostly reduction (or no effect) in percutaneous permeation or systemic absorption, Phospholipids are able to diffuse into the subcutaneous (SC), and the interactions and enhancer effects of liposomes on the SC are based on the lipid mixing of liposomal phospholipids with lipid bilayers of the skin [17].
[adsense:468x15:2204050025]
Phospholipids in liposomal systems can disrupt the bilayer fluidity in the SC, decreasing the barrier properties of the skin. Moreover, some investigators report that phospholipids in liposomes may mix with the SC lipids creating a lipid-enriched environment [18]. This lipid depot in the skin is preferred by lipophilic drugs, resulting in enhanced skin uptake. In some cases, phospholipids themselves can be the solubilizers to increase the solubility of lipophilic drugs such as indomethacin and miconazole [19]. These results suggested that conventional liposomes were useful for topical dermal delivery of these drugs. Several recent studies suggested other applications and confirmed improved skin deposition [20-23]. The extent of interaction between lipid vesicles and skin was highly dependent on the lipid composition of the liposomes, confocal microscopy studies showed that intact liposomes were not able to penetrate into granular layers of the epidermis [24]. Liposomes should exhibit their enhancing effect on skin in the presence of organic solvents such as propylene glycol, tetraglycol and ethanol. The concentration of phospholipids and the presence of unsaturated fatty acids in phospholipids are also important factors affecting transdermal flux of drugs [25- 27].
The rationale for using vesicles in dermal and transdermal drug delivery is many folds. Vesicles might: (a) act as drug carriers to deliver entrapped drug molecules into or across the skin;(b) act as penetration enhancers owing the penetration of the individual lipid components into the stratum corneum and subsequently the alteration of the intercellular lipid lamellae within this skin layer; (c) serve as a depot for sustained release of dermally active compounds;(d) serve as a rate-limiting membrane barrier for the modulation of systemic absorption, hence providing a controlled transdermal delivery system.
To obtain a sound understanding of the mechanism of the action of vesicles and to select the most appropriate drug compounds to be delivered by vesicles (Fig2.). If vesicles act as carrier systems, they might be able to transport large molecular weight drugs, such as proteins into the skin or even into the systemic circulation. If they act as penetration enhancers, however, the main mode of action is a perturbation of the lipid organization in the stratum corneum, thereby increasing the transport rate across the skin. The latter is only efficient for low molecular weight drugs. One of the most important characteristics of drug carrier systems is that drug and carrier should permeate along the same route across the skin. In addition the vesicle material profile and the active compound profile in stratum corneum should be very similar [2].
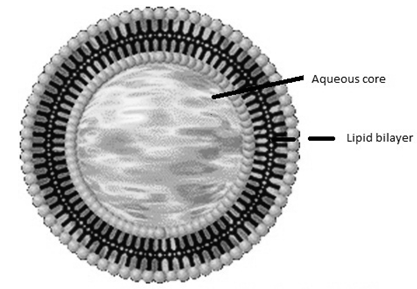
Figure. 1: Schematic spherical vesicles with a phospholipid bilayer
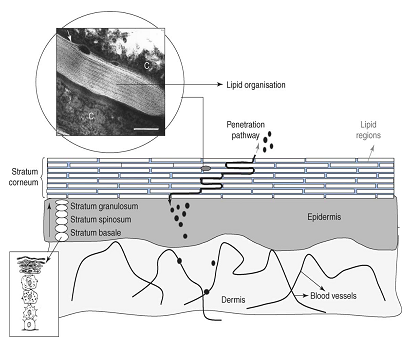
Figure 2. A schematic drawing of a skin cross-section [28]. The skin is composed of a dermis and an epidermis. In the basal layer of the epidermis cells proliferate. Upon leaving the basal layer cells start to differentiate and migrate in the direction of the skin surface. At the interface between stratum granulosum–stratum corneum final differentiation occurs, during which the viable cells are transformed into dead keratin filled cells (corneocytes). The corneocytes are embedded in lipid lamellar regions. Substances permeate mainly along the tortuous pathway in the intercellular lamellar regions. The thickness of the stratum corneum is approximately 15 mm. C = corneocyte filled with keratin. Bar = 100 nm.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Deformable lipid vesicles:
Intensive investigations led to the introduction and development, of a new class of highly deformable (elastic or ultraflexible) liposomes that have been termed Transfersomes®. While conventional liposomes were reported to have mainly localizing or rarely transdermal effects, deformable liposomes were reported to penetrate intact skin, carrying therapeutic concentrations of drugs, but only when applied under non-occluded conditions. Deformable liposomes (Transfersomes®) are the first generation of elastic vesicles introduced by Cevc and Blume (1992) [29]. Transfersomes are highly efficient edge activator -based ultra flexible vesicles capable of, noninvasively, trespassing skin by virtue of their high, self-optimizing deformability. They consist of phospholipids and an edge activator. Transfersomes for enhanced transepidermal delivery of Diclofenac sodium, an edge activator is often a single chain surfactant, having a high radius of curvature, which destabilizes lipid bilayers of the vesicles and increases deformability of the bilayers [30]. They are liquid-state vesicles with a highly deformable membrane [31, 32] which permits their easy penetration through skin pores much smaller than the vesicles’ size. They have been proven to be superior to conventional gel-state and even liquid-state vesicles in terms of both [33, 34], the enhancement of drug permeation and interactions with human skin [35, 36]. The ultra-flexible carrier’s hydration sensitivity and its unique driving force create an unprecedented opportunity to control the depth of the carriers’ migration, by selecting different drug dose and/or carrier dose per area (Fig.3). Transfersomes can effectively protect the drug against undesired rapid clearance into cutaneous blood vessels and are capable of retaining the drug long enough, on, in and below the skin barrier. Furthermore, they can cross the stratum corneum independent of drug concentration. Occluded passive penetration improved estradiol skin penetration from liposomes relative to control. Iontophoretic studies revealed the superiority of ultradeformable vesicles regarding drug skin penetration and deposition compared to traditional liposomes [37, 38]. Iontophoresis with ultradeformable liposomes improved estradiol transepidermal penetration in vitro [39]. The improvement in drug penetration was suggested to be due partly to the high deformability of the vesicular membrane. This vesicular elasticity would have resulted in penetration of some vesicles through the skin under the stress imparted by the applied electric current [40]. Penetration enhancers were able to give more deformable vesicles than conventional liposomes with a good drug entrapment efficiency and stability [41]. In vitro skin penetration data showed that Penetration Enhanced Vesicles were able to give a statistically significant improvement of minoxidil deposition in the skin in comparison with classic liposomes and penetration enhancer-containing drug ethanolic solutions without any transdermal delivery [42, 43].
Deformable liposomes were also reported to improve both in vitro skin permeation and deposition of cyclosporin A [44], methotrexate [45], and melatonin [46]. They significantly improved ketotifen skin delivery, with greater improvement of ketotifen skin deposition than improvement of ketotifen skin permeation, hence were suggested to be more useful for dermal than for transdermal delivery of ketotifen [47]. They were also reported to improve only skin deposition of 5-fluorouracil [48] and dipotassium glycyrrhizinate [49], hence were considered only useful for dermal delivery of these drugs. Reported success of deformable liposomes to deliver macromolecules and proteins such as insulin through intact human skin with efficiency comparable with subcutaneous administration led to their introduction as possible carriers for non-invasive gene delivery and transcutaneous immunization. Investigated deformable cationic liposomes, prepared using a cationic lipid, 1, 2-dioleoyl-3-trimethylammoniumpropane, and sodium cholate, as gene delivery system. In vitro transfection efficiency of plasmid DNA was assessed by the expression of green fluorescent protein (GFP) [50]. This formulation was capable of transfecting several cell lines. It was also tested for in vivo transfection efficiency and its retention time within the organs, by applying the complexes on hair-removed dorsal skin of mice, non-invasively. It was found that genes were transported into several organs for 6 days once applied on intact skin, suggesting promising properties for non-invasive gene delivery. In another study, deformable liposomes prepared using egg-phosphatidylcholine could also deliver genetic materials (GFP) into mice transdermally [51].

Figure 3. The schematic drawing of skin permeation of lipid vesicles (A) lipid vesicles with permeation enhancer (B) and flexible vesicles (C); the direct transfer of drug is due to the interaction of skin lipids and vesicles; flexible vesicles permeation due to the hydration or osmotic gradient.
Ethosomes:
Vesicular carriers with promising results in dermal and transdermal drug delivery are ethosomes. Discovered by Touitou et al. (1997) [52], ethosomes contain phospholipids, alcohol (ethanol/isopropyl alcohol) in relatively high concentration and water. The use of high ethanol content was first described for ethosomes. Due to the inter-digitation effect of ethanol on lipid bilayers, it was believed that high concentrations of ethanol are damaging to liposomal formulations. However, ethosomes which are novel permeation-enhancing lipid vesicles embodying high concentration (20–45%) of ethanol were developed and investigated. Ethosomes are most commonly prepared by the lipids and the drug is dissolved in ethanol. The aqueous component is added slowly in a fine stream at constant rate in a well-sealed container with constant mixing. Mixing is then continued for additional few minutes. These vesicular carriers having the size range of few nanometers to microns are claimed to be most suited for the transdermal drug delivery of bioactives. Unlike classical liposomes, ethosomes were shown to permeate through the stratum corneum barrier and were reported to possess significantly higher transdermal flux in comparison to liposomes. The exact mechanism for better permeation into deeper skin layers from ethosomes is still not clear however the synergistic effects of combination of phospholipids and high concentration of ethanol in vesicular formulations have been suggested to be responsible for deeper distribution and penetration in the skin lipid bi-layers [53-56]. Ethosomal carriers have been shown to be effective permeation enhancers. The delivery of biologically active agents has been well reported. Dayan and Touitou reported the facilitation of probe penetration to a greater skin depth with ethosomal carriers, and also relatively high fluorescence intensity, compared to a hydroethanolic solution and conventional liposomes, using various probes of different nature, like D-289 and Rhodamine Red [57].
Recently studies reported for the delivery of anti-HIV/AIDS drugs via ethosomes. The encapsulation of AZT in ethosomes and compared its transdermal potential with liposomes and hydroethanolic and ethanolic solutions of the drug [58]. Intercellular and intracellular drug delivery of lamivudine was improved from ethosomes using visualization techniques and cell line study. The results concluded that ethosomes could be advantageous in terms of transdermal drug delivery of the anti-HIV/AIDS agents [59, 60].
Table 1. Some recently reported studies investigating efficiency and applications of ethosomes as carriers for transdermal delivery of drugs
|
Drug |
Investigation |
Results |
|
Testosterone |
Pharmacokinetics |
AUC was about 64% greater with ethosomes than with commercial gel [61]. |
|
Cannabidiol |
Suppression of carrageenan-induced aseptic paw edema (anti-inflammatory action) |
Development of edema was prevented entirely only in pretreated (ethosomal patch) group of mice. Delta in paw thickness of pretreated mice was statistically different from that of the non-pretreated mice starting from 1 h post-carrageenan injection and lasting until the end of the inflammation course [62] |
|
Erythromycin |
In vivo antibacterial efficiency |
Ethosomal erythromycin resulted in complete inhibition of infection while hydroethanolic erythromycin solution caused deep dermal and subcutaneous abscesses within 5 days after challenge [63]. |
|
Ammonium glycyrrhizinate |
Suppression of chemically induced erythema (anti-inflammatory action) |
Ethosomes reduced the erythema more rapidly with respect to drug solutions. Ethosomes also showed sustained effect [64]. |
|
Minoxidil
|
In-vitro permeation through Rat abdominal skin |
Addition of cholesterol significantly improved skin delivery from ethosomes [65]. |
|
Azelaic acid |
Diffusion through synthetic membranes |
Ethosomes having the highest ethanol concentration released the drug more rapidly [66]. |
|
Ketotifen |
Diffusion across Rabbit pinna skin |
Ethosomes improved cumulative drug permeated [47] |
|
hepatitis B surface antigen |
in vitro qualitative and quantitative uptake by human dendritic cells |
Ethosomes coupled with their skin navigating potential, make it an attractive vehicle for development of a transcutaneous vaccine against hepatitis B in preference to elastic liposomes [67]. |
|
Methotrexate |
Dermal and transdermal delivery of an anti-psoriatic agent |
Ethosomes with some visual penetration pathways and corneocytes swelling, a measure of retentive nature of formulation [68]. |
|
5-aminolevulinic Acid |
encapsulated liposome versus ethosome for skin delivery |
the penetration ability of ethosomes was greater than that of liposomes [69] |
|
Melatonin |
skin permeation |
Formulation revealed a greater mobility of skin lipids on application of ethosomes as compared to that of ethanol or plain liposomes [70]. |
Invasomes:
Vesicles, have been intensively studied as drug carrier systems for dermal and transdermal administration of drugs. Since liquid-state vesicles have been proven to be superior to rigid gel-state vesicles in terms of enhanced drug penetration, and elastic vesicles have been shown to be superior to conventional gel-state and even liquid-state vesicles in terms of interactions with human skin and enhanced drug penetration a series of liquid-state vesicles with elastic membranes were developed [71].
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Invasomes composed of phosphatidylcholine, ethanol and terpenes [72]. One proposed mechanism of the penetration enhancing ability of elastic vesicles is that vesicles or vesicle constituents disorganise and disrupt intercellular lipid lamellae forming channel-like penetration pathways, through which drug molecules penetrate. The second proposed theory is that intact vesicles penetrate into SC through pre-existing channels with low penetration resistance, which is also supported by Cevc et al. Honeywell-Nguyen et al. demonstrated the partitioning of intact vesicles into deeper layers of SC. Terpenes, as constituents of invasomes, present potent penetration enhancers and they have been shown to enhance the percutaneous absorption of midazolam [73], indomethacin, lorazepam, klonazepam [74], haloperidol [75], nicardipine, carbamazepine, tamoxifen [76] and many other drugs. Investigations employing differential scanning calorimetry and X-ray diffraction revealed that terpenes increase drug permeation by disrupting lipid packaging of SC and/or disturbing the stacking of the bilayers [77-79]. Ethanol, which has been also added to invasomes, presents an efficient penetration enhancer [80-83] and a synergistic effect of liposomes and ethanol has been described in the literature [84]. Invasomes could be a promising tool for delivering the highly hydrophobic photosensitizer temoporfin to the skin, which would be advantageous for the photodynamic therapy of cutaneous malignant (basal-cell carcinoma) or nonmalignant diseases [85].
Preparation of lipid vesicular system:
All the lipid vesicles were prepared by a conventional rotary evaporation method [86, 87]. Briefly, the appropriate weights of lipid or lipids were dissolved in methanol/chloroform solution (1:2, v/v) in round bottom flask. Thin lipid films were obtained by removing the organic solvents under vacuum condition (500 mbar 10 min, 200 mbar 10 min, 100 mbar 10 min, 35 mbar 1 hr) at a temperature more than transition temperature of lipid with a rotary evaporator. The traces of solvent were removed from the deposited lipid film under vacuum overnight. The resulted dry lipid films on the inside wall of round bottom flask were hydrated and dispersed with different hydration systems like phosphate buffered saline corresponding to all formulations at room temperature. The obtained macroscopically homogenous solution was sonicated for totally 15 min in 3 cycles (5 min for each cycle and 5 min pause among these cycles) with a sonication ice-water bath. Then these suspensions were extruded through a series of 0.45, 0.22-μm polycarbonate membrane many times to produce liposomes of the desired size with the help of a Hamilton-Bonaduz extruder [88].
Characterization of different lipid vesicular systems:
Vesicles size and surface morphology:
Transmission electron microscope (TEM) was used as a visualizing aid for lipid vesicles. Scanning electron microscopy (SEM) is also conducted to characterize the surface morphology of the vesicles. The size distribution is determined, by Dynamic Light Scattering (DLS) technique using a computerized Autosizer inspection system. For vesicles size measurement, vesicular suspension is mixed with the appropriate buffer medium and a drop of vesicle dispersion is applied to a carbon film-covered copper grid and is stained with a 1% phosphotungstic acid. Then, samples are examined with a transmission electron microscope at an accelerating voltage of 80 kV. All particle sizes and zeta potentials are measured at room temperature. In order to evaluate the stability of ethosomes during storage, the size and zeta potential of vesicles are also monitored after being stored at 4 ?C [46].
Determination of the drug entrapment efficiency:
A centrifugation method was used to separate the incorporated drug in the free form. Vesicle suspensions were centrifuged. Following centrifugation, the supernatant and pellet were separated, and the concentration of drug was analyzed by high-performance liquid chromatography (HPLC) or spectrophotometrically [81].
Deformation index determination:
Comparative measurement of deformability of liposome bilayer with different penetration enhancers is generally carried out by extrusion measurement. The vesicle dispersion extruded at constant pressure through polycarbonate filters of definite pore size using an extrusion device. The deformability of vesicle is expressed in terms of deformation index (DI).
DI= j(d0/p)k (1/|d1 - d0| )
Where J is the amount of suspension recovered after extrusion, d0 and d1 are the mean diameter of vesicles before and after extrusion, p is the pore size of the membrane, and k is an amplification factor [34].
In vitrodrug release (flux studies):
The permeation of DS-bearing Transfersomes through an artificial cellophane membrane was performed in Franz-type diffusion cells with a diffusion area of 1.77cm2 [90]. The receptor medium was phosphate buffer which was constantly stirred at 100rpm with a small magnetic bar. The receptor compartment was maintained at 37±0.2 ?C by a circulating water jacket. An equivalent amount was placed in the donor compartment. Samples were withdrawn from the receptor compartment via the sampling port at different time intervals to 24 h, and immediately replaced with an equal volume of fresh receptor solution. All samples were analyzed for drug content spectrophotometrically or HPLC. The obtained data were kinetically treated to determine the order of release. The flux at 24 h (J) was assessed and the release profile curves were constructed for all formulae [60].
I = Amount of permeated drug/ Time x Area of release membrane
In vivo, ex vivopermeation study:
The assessment of percutaneous permeation of molecules is one of the main steps in the initial design and later in the evaluation of dermal or transdermal drug delivery systems. The literature reports numerous ex vivo, in vitro and in vivo models used to determine drug skin permeation profiles and kinetic parameters, some studies focusing on the correlation of the data obtained using these models with the dermal/transdermal absorption in humans. In- vitro permeation studies to clinical performance, presenting various experimental models used in dermal/transdermal research, including the use of excised human or animal skin, cultured skin equivalents and animals (rat, mice, rabbit, pig etc.) [91].
Visualization of skin penetration using confocal laser scanning microscopy (CLSM):
CLSM has provided a significant tool with which to visualize skin structure and the localization of fluorescent probes within the tissue. Because of its nondestructive nature, and the fact that little or no sample preparation is necessary, CLSM offers a reasonably faithful representation of reality with few artifacts. The potential to recreate three-dimensional visualization of the tissue is another significant advantage relative to other microscopic techniques. CLSM provides complementary image information to that obtained from other conventional microscopic and histological methods. The major contribution of CLSM in the topical/transdermal field to-date have been mechanistic, particularly in terms of revealing preferred penetration pathways following, the use of different delivery technologies. Nevertheless, there remain important limitations of CLSM. First, only a restricted range of fluorophores are available for imaging and attachment of these markers, for example, to a dry of interest may change significantly the permeability behaviour (rate, extent, route of transport, etc.). Second, the technique reveals at best only semi-quantitative information, and no approach to calibrate the fluorescence intensities observed has yet been demonstrated. Third, the images obtained are static views of reality captured at a particular point in time; thus, does a strongly fluorescent region imply a key pathway through which a large fraction of the total transport is occurring, or does it suggest an area where the fluorophore has become tightly bound and/or immobilized [92].
CURRENT AND FUTURE DEVELOPMENTS:
The ability of lipid vesicles consisting of components other than phospholipids and cholesterol or their semisynthetic derivatives [93] to enhance the encapsulation of bioactive substances provides new promising perspectives for establishing new, efficient and stable carriers for drug delivery. The potential of lipid vesicles for transdermal iontophoretic drug delivery was first reported by Vutla et al. (1996) [94]. Some studies were followed, investigating such approach, using either traditional or deformable liposomes [95-97]. The exact benefits remain unclear, as a result of paucity of reported investigations; however, it appears that combining iontophoresis with lipid vesicles facilitates drug delivery to deeper layers of skin including enhanced trans-follicular delivery. Studies will continue to further investigate such a promising approach.
A new lipid vesicular system, the propylene glycol-embodying liposomes (PG-liposomes), composed of phospholipid, propylene glycol and water, was recently introduced, developed and investigated, as a carrier for skin delivery of drugs. PG-liposomes of the local anesthetic, cinchocaine, showed high entrapment efficiency and were relatively more stable than other liposomal formulations, including deformable liposomes and ethosomes. In vivo skin deposition studies, carried out using rabbit dorsal skin, indicated that cinchocaine PG-liposomes were superior, as carriers for skin delivery, relative to other liposomal formulations. PG-liposomes showed improved skin deposition, even relative to deformable liposomes and ethosomes, suggesting that PG-liposomes, recently developed, may have promising future as carriers for skin delivery of drugs [47]. Another approach that could increase the photoprotective effect against UV radiation comprises targeted delivery of α tocoperol into the deeper skin layers and across the cell membranes. For this purpose, ethosomal vitamin E compositions were designed and tested [98, 99]. Ethosomes are phospholipid soft vesicles composed of safe and natural components approved for pharmaceutical and cosmetic use.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
CONCLUSION:
Extensive studies are reported to enhance efficiency of lipid vesicles on dermal or transdermal drug delivery. However, most of the studies focused on in vitro or laboratory experiments. Confirmation of lipid vesicles for clinical use may extend their applicability. Although liposomes are theoretically thermodynamically stable, conquering the stability problem during storage is an important issue. The instability of liposomes can cause leakage of encapsulated drugs from the vesicles and aggregation and/or fusion to form larger vesicles. The classic nano-sized vesicles may not exhibit their benefits after aggregation. Advanced attempts have been successfully applied in the laboratory, many problems need to be overcome and investigations may be modified for the commercial patents and products in the future. The valid preparation procedures from laboratory scale to mass production are also important for resolving these realistic difficulties is the challenge and mission for future development of new products and formulations of lipid vesicular systems.
ACKNOWLEDGEMENTS:
The authors thank Dr. M. S. Ranawat and Dr. C.S. Chauhan, B. N. College of Pharmacy, Udaipur, Rajasthan for their technical assistance, valuable suggestions and comments during the preparation of this review article.
REFERENCES:
[1] B.W. Barry, Eur. J. Pharm. Sci., 2001,14, 101–114.
[2] P. L. Honeywell-Nguyen, J.A. Bouwstra, Drug Discov. Today: Technol., 2005, 2, 67–74.
[3] A.Williams, Transdermal and Topical Drug Delivery, Pharmaceutical Press, London, 2000.
[4] H. Schaefer, and T. E. Redelmeier, Skin barrier. In Principles of percutaneous absorption, Karger, 1996.
[5] G. M. El Maghraby, A. C. Williams, B.W. Barry, J. Pharm. Pharmacol., 2006, 58, 415–429.
[6] M. Mezei, V. Gulasekharam, Life Sci., 1980, 26, 1473–1477.
[7] E. Touitou, M. Alkabes, N. Dayan, Pharm. Res., 1997, S14, 305–306.
[8] N. Dayan, E. Touitou, Biomaterials., 2000, 21, 1879–1885.
[9] E. Touitou, N. Dayan, L. Bergelson, B. Godin, M. Eliaz, J. Control. Release., 2000, 65, 403–418.
[10] E. Touitou, B. Godin, C. Weiss, Drug Dev. Res., 2000, 50, 406–415.
[11] J.K. Vasir, M. K. Reddy, V.D. Labhasetwar, Curr Nanosci., 2005, 1, 47-64.
[12] M.C. Woodle, D. Papahadjopoulos, Methods Enzymol., 1989, 171, 193-217.
[13] J. Gubernator, M. Stasiuk, A. Kozubek, Biochim. Biophys. Acta., 1999, 1418, 253- 260.
[14] A. Kozubek, M. Nietubyc, A.F. Sikorski, Z. Naturforsch., 1992, 47c, 41-46.
[15] J. Gubernator, PhD Thesis, University of Wroclaw, (Wroclaw, 1998).
[16] J. Gubernator, A. Kozubek, Chem. Phys. Lipids., 1998, 94, 177.
[17] M. Kirjavainen, J. Monkkonen, M. Saukkosaari, R. Valjakka- Koskela, J. Kiesvaara, A. Urtti, J. Control. Release., 1999, 58, 207-14.
[18] C. Valenta, M. Wanka, J. Heidlas, J. Control. Release., 2000, 63, 165-73.
[19] M. Fujii, K. Shiozawa, Y. Watanabe, M. Matsumoto, Int. J. Pharm., 2001, 222, 57-64.
[20] L.S. Ferreira, G.A. Ramaldes, E.A. Nunan, L.A. Ferreira, Drug Dev. Ind. Pharm., 2004, 30, 289–296.
[21] S. Kitagawa, M. Kasamaki, Chem. Pharm. Bull., 2006, 54, 242–244.
[22] C. Puglia, D. Trombetta, V. Venuti, A. Saija, F. Bonina, J. Pharm. Pharmacol., 2004, 56, 1225–1232.
[23] E. Ramon, C. Alonso, L. Coderch, A. de la Maza, O. Lopez, J.L. Parra, J. Notario, Drug Deliv., 2005, 12, 83–88.
[24] M. Kirjavainen, A. Urtti, I. Jaaskelainen, T.M. Suhonen, P. Paronen, R. Valjakka- Koskela, J. Kiesvaara, J. Monkkonen, Biochim. Biophys. Acta., 1996, 1304, 179–189.
[25] C. Valenta, M. Wanka, J. Heidlas, J. Control. Release., 2000, 63, 165-73.
[26] M. Fujii, K. Shiozawa, Y. Watanabe, M. Matsumoto, Int. J. Pharm ., 2001, 222, 57-64.
[27] R. Valjakka-Koskela, M. Kirjavainen, J. Monkkonen, A. Urtti, J. Kiesvaara, Int. J. Pharm. 1998, 175, 225-30.
[28] J. A. Bouwstra, Prog. Lip. Res., 2003, 42, 1–36.
[29] G. Cevc, Crit. Rev. Ther. Drug Carrier Syst., 1996, 13, 257–388.
[30] G. M. El Zaafarany, G. A.S. Awad, S.M. Holayel, N. D. Mortada, Int. J. Pharm., 2010, 397, 164–172.
[31] G. Cevc, G. Blume, Biochim. Biophys. Acta., 2003, 1614, 156–164.
[32] Cevc, G., Vierl, U., Mazgareanu, S., 2008. Functional characterisation of novel analgesic product based on self-regulating drug carriers. Int. J. Pharm. 360, 18–28.
[33] J. Montanari, D.I. Roncaglia, L.A. Lado, M.J. Morilla, E.L. Romero, Int. J. Pharm., 2009, 372, 184–190.
[34] M. Manconi, S. Mura, C. Sinico, Fadda, A.O. Vila, F. Molina, Physicochem. Eng. Asp., 2009, 342, 53–58.
[35] N. Dragicevic-Curic, S. GrAfe, B. Gitter, S. Winter, A. Fahr, Int. J. Pharm., 2010, 384, 100–108.
[36] B.A.I. Van den Bergh, P.W. Wertz, H.E. Junginger, J.A. Bouwstra, Int. J. Pharm., 2001, 217, 13–24.
[37] E.A. Essa, M. C. Bonner, B. W. Barry, J. Control. Release., 2004, 95, 535– 546.
[38] E.A. Essa, M.C. Bonner, B.W. Barry, Int. J. Pharm., 2002, 240, 55– 66.
[39] G.M.M. El-Maghraby, A.C. Williams, B.W. Barry, Int. J. Pharm., 2000, 196, 63– 74.
[40] G.M.M. El Maghraby, A.C. Williams, B.W. Barry, J. Pharm. Pharmacol., 2001, 53, 1311 – 1322.
[41] Williams, A.C., Barry, B.W., In: Touitou, E., Barry, B.W. (Eds.), Enhancement in Drug Delivery. (CRC Press, Boca Raton), 2007, 242–243.
[42] S. Mura, F. Pirot, M. Manconi, F. Falson, A.M. Fadda, J. Drug Target., 2007, 15, 101–108.
[43] S. Mura, M. Manconi, C. Sinico, D. Valenti, A.M. Fadda, Int. J. Pharm., 2009, 380, 72–79.
[44] J. Guo, Q. Ping, G. Sun, C. Jiao, Int. J. Pharm., 2000, 194, 201–207.
[45] M. Trotta, E. Peira, M. E. Carlotti, M. Gallarate, Int. J. Pharm., 2004, 270, 119– 125.
[46] V. Dubey, D. Mishra, A. Asthana, N. K. Jain, Biomaterials., 2006, 27, 3491–3496.
[47] M. M Elsayed,O. Y. Abdallah, V. F. Naggar, N. M. Khalafallah, Pharmazie., 2007, 62, 133–137.
[48] G. M. El Maghraby, A. C. Williams, B. W. Barry, J. Pharm. Pharmacol., 2001, 53, 1069–1077.
[49] M. Trotta, E. Peira, F. Debernardi, M. Gallarate, Int. J. Pharm., 2002, 241, 319–327.
[50] A. Kim, E. H. Lee, S. H. Choi, C. K. Kim, Biomaterials., 2004, 25, 305–313.
[51] E. H. Lee, A. Kim, Y. K. Oh, C. K. Kim, Biomaterials., 2005, 26, 205–210.
[52] E. Touitou, N. Dayan, L. Bergelson, B. Godin, M. Eliaz, J. Control. Release., 2000, 65, 403–418.
[53] US patent: Inventor E. Touitou, Composition of applying active substance to or through the skin, 5 540 934 (1996).
[54] US patent: Inventor E. Touitou, Composition of applying active substance to or through the skin, 5 716 638 (1998).
[55] E. Touitou, B. Godin, N. Dayan, C.Weiss, A. Piliponsky, F. Levi-Schaffer, Biomaterials., 2001, 22, 3053–3059.
[56] V. Dubey, D. Mishra, M. Nahar, N.K. Jain, Drug Deliv., 2007, 4, 579–593.
[57] N. Dayan, E. Touitou, Biomaterials., 2000, 21, 1879–1885.
[58] S. Jain, R.B. Umamaheshwari, D. Bhadra, N.K. Jain, Indian J. Pharm. Sci., 2004, 66, 72–81.
[59] S. Jain, A.K. Tiwary, B. Sapra, N.K. Jain, AAPS Pharm. Sci. Tech., 2007, 8, E1–E9.
[60] U. Gupta, N. K. Jain, Advanced Drug Delivery Reviews., 2010, 62, 478–490.
[61] D. Ainbinder, E. Touitou, Drug Deliv., 2005, 12, 297–303.
[62] M. Lodzki, B. Godin, L. Rakou, R. Mechoulam, R. Gallily, E. Touitou, J. Control. Release., 2003, 93, 377–387.
[63] B. Godin, E. Touitou, Curr. Drug Deliv., 2005, 2, 269–275.
[64] D. Paolino, G. Lucania, D. Mardente, F. Alhaique, M. Fresta, J. Control. Release., 2005 106, 99–110.
[65] J.M. Lopez-Pinto, M. L. Gonzalez-Rodriguez, A. M. Rabasco, Int. J. Pharm., 2005, 298, 1–12.
[66] E. Esposito, E. Menegatti, R. Cortesi, J. Cosmet. Sci., 2004, 55, 253–264.
[67] D. Mishra, P. K. Mishra, S. Dabadghao, V. Dubey, M. Nahar, N. K. Jain, Nanotech. Bio. Med. 2010, 6, 110–118.
[68] V. Dubey, D. Mishra, T. Dutta, M. Nahar, D.K. Saraf, N.K. Jain, J.Control. Release., 2007, 123, 148–154.
[69] Y. Fang, Y. Tsai, P. Wu, Y. Huang, Int. J. of Pharm., 2008, 356, 144–152.
[70] V. Dubey, D. Mishra, N.K. Jain, Eur. J. Pharm. and Biopharm., 2007, 67, 398–405.
[71] N. Dragicevic-Curic, D. Scheglmann, V. Albrecht, A. Fahr, J. Control. Release., 2008, 127, 59–69.
[72] D.D. Verma, S. Verma, K.J. McElwee, P. Freyschmidt-Paul, A. Fahr, Eur. J. Dermatol., 2004, 14, 332–338.
[73] Y. Ota, A. Hamada, M. Nakano, H. Saito, Drug Metab. Pharmacokin., 2003, 18, 261–266.
[74] T. Ogiso, M. Iwaki, P. Tsuyoshi, J. Pharm. Sci., 1995, 84, 482–488.
[75] C. Puglia, F. Bonina, G. Trapani, M. Franco, M. Ricci, Int. J. Pharm., 2001, 228, 79–87.
[76] H.K. Vaddi, P.C. Ho, Y.W. Chan, S.Y. Chan, J. Control. Release., 2002, 81, 121–133.
[77] A.F. El-Kattan, C.S. Asbill, N. Kim, B.B. Michniak, Int. J. Pharm., 2001, 215, 229–240.
[78] P.A. Cornwell, B.W. Barry, C.P. Stoddart, J.A. Bouwstra, J. Pharm. Pharmacol., 1994, 46, 938–950.
[79] P.A. Cornwell, B.W. Barry, J.A. Bouwstra, G.S. Gooris, Int. J. Pharm., 1996, 127, 9–26.
[80] K.S. Bhatia, J. Singh, Int. J. Pharm., 1999, 180, 235–250.
[81] D. Kobayashi, T. Matsuzawa, Y. Sugibayashi, Y. Morimoto, M. Kimura, Pharm. Res., 1994, 11, 96–103.
[82] S. Tata, N. Weiner, G. Flynn, J. Pharm. Sci., 1994, 83, 1508–1510.
[83] V. Srinivasan, J. Pharm. Sci., 1990, 79, 558–591.
[84] N. Dragicevic-Curic, D. Scheglmann, V. Albrecht, A. Fahr. Biointerfaces., 2009, 70, 198–206.
[85] N. Dragicevic-Curic, D. Scheglmann, V. Albrecht, A. Fahr. J. Control. Release., 2008, 127, 59–69.
[86] E. R. Bendas, M. I. Tadros, AAPS Pharm. Sci.Tech., 2007, 8, E107.
[87] K. B. Ita, , J. Du Preez, M.E. Lane, J. Hadgraft, J. du Plessis, , J. Pharm. Pharmacol., 2007, 59, 1215–1222.
[88] R. C. MacDonald, R. I. MacDonald, B. P. M. Menco, K. Takeshita, N. K. Subbarao, L. R. Hu, Biochim. Biophys. Acta., 1991, 1061, 297–303.
[89] M. Chen, X. Liu, A. Fahr, Int. J. Pharm., 2011, 408, 223–234.
[90] P.N. Gupta, V. Mishra, A. Rawat, P. Dubey, S. Mahor, S. Jain, D. P. Chatterji, S.P. Vyas, Int. J. Pharm., 2005, 293, 73–82.
[91] B. Godin, E. Touitou, Adv. Drug. Del. Rev., 2007, 59, 1152–1161.
[92] R. Alvarez-Roma´n, A. Naik, Y.N. Kalia, H. Fessi, R.H. Guy, Eur. J. Pharm. and Biopharm., 2004, 58, 301–316.
[93] A. Kozubek, J. Gubernator, E. Przeworska and M. Stasiuk, Acta. Bio. Polonica., 2000, 47, 639–649.
[94] N. B. Vutla, G.V. Betageri, A. K. Banga, J. Pharm. Sci., 1996, 85, 5–8.
[95] J. Y. Fang, K.C. Sung, H. H. Lin, C.L. Fang, J. Control. Release., 1999, 60, 1–10.
[96] R. R. Boinpally, S. L. Zhou, G. Devraj, P. K. Anne, S. Poondru, B. R. Jasti, Int. J. Pharm., 2004, 274, 185–190.
[97] Han, M. Kim, J. Kim, Exp. Dermatol., 2004, 13, 86–92.
[98] E. Touitou, B. Godin, J. Appl. Cosmetol., 2006, 24, 139-47.
[99] S. Lavy, M. Sc. Thesis, The Hebrew University of Jerusalem (Jerusalem, Israel, 2002).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








