

{ DOWNLOAD AS PDF }
ABOUT AUTHORS
Aliyu Hamidu Ahmed1, Yusuf Ibrahim Alkali2
1Department of Pharmacognosy and Ethno pharmacy, Faculty of Pharmaceutical Sciences
Usmanu Danfodiyo University, Sokoto- Nigeria
2Department of Pharmacology and Toxicology, Usmanu Danfodiyo University, Sokoto- Nigeria.
ABSTRACT
Pharmacokinetics and toxicity profile along with efficacy are the major determinants for successful drug development. This study was carried out to determine the pharmacokinetic profile, potential biological activities and toxicity of diospyrin, lupeol and plumbagin using in silico approaches. The Swiss ADME tool was used to calculate the molecular properties of the ligands based on Lipinski’s rule of five (5).All ligands in the present study satisfied the rule. Using the Swiss ADME tool, the pharmacokinetic profile of the compounds was evaluated. Protox-II server was used to predict the organ toxicities and toxicological end points of the ligands and their LD50. Plumbagin is found to have both mutagenicity and carcinogenicity. Lupeol and diospyrin are reported to be immunotoxic. Lupeol has LD50 of 2000mg/kg. Diospyrin and plumbagin have 16mg/kg. Swiss target prediction server was used to identify the various potential target. The target prediction suggests that plumbagin and lupeol have high preference for Microtubule-associated protein tau (MAPT). The best target for diospyrin was Aurora kinase A. Molecular docking study was conducted using AutoDock vina in The Python Prescription (PyRx) 0.8 virtual screening tool. Plumbagin and lupeol were docked against Microtubule associated protein tau. The dockings scores based on binding energy were; plumbagin -33.8 (kcal/mole) and lupeol -44.7 (kcal/mole). Diospyrin showed a binding energy of -10.7 (kcal/mole) against Aurora A kinase. Results in this study suggest that diospyrin may serve as an important aurora kinase inhibitor while lupeol and plumbagin may be useful in treatment of Alzheimer’s disease.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-2651
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 7, Issue 03 Received On: 07/01/2019; Accepted On: 16/02/2019; Published On: 01/03/2019 How to cite this article: Ahmed, A. and Alkali, Y. 2019. In silico Pharmacokinetics and Molecular Docking Studies of Lead Compounds Derived from Diospyros Mespiliformis. PharmaTutor. 7, 3 (Mar. 2019), 31-37. DOI:https://doi.org/10.29161/PT.v7.i3.2019.31 |
INTRODUCTION
Drug discovery is an extreme multi-step and interdisciplinary endeavor that follows a sequential process starting with target and lead discovery, followed by lead optimization and pre-clinical in vitro and in vivo studies to determine the suitability of such compounds through a number of pre-set criteria for initiating clinical development (Pugazhendhi et al., 2013). The high cost and consuming nature of these processes called for the development of in silico approach which was found to be cost effective and less time consuming (Pakomwit et al., 2015). Pharmacokinetics and toxicity profile along with efficacy are the major determinants for successful drug development (Moroy et al., 2012). Using the in silico approach, the pharmacokinetics and toxicity studies of compounds are done before they are evaluated for biological activity (Van de Waterbeemd, 2002). Poor pharmacokinetic profile and toxicity are the main reasons for late-stage failures in drug discovery. Therefore it is necessary that these areas should be considered at the early stages in the drug discovery process (Pelkonen et al., 2011). Computer designed models are a valid alternative to experimental procedures for prediction of pharmacokinetics (Dahlin et al., 2015).
Molecular Docking is an important method in molecular biology and computer-aided drug design. Ligand- protein docking helps predict the binding mode of a Ligand and Protein of known 3-dimensional structures (Cross et al., 2009). Molecular Docking is frequently used to predict the binding orientation of drug candidates to their protein targets in order to predict the affinity and activity of the small molecule. Hence docking plays an important role in the rational design of drugs (Kitchen et al., 2004) .The aim of molecular docking is to achieve an optimized conformation for both the protein and ligand and relative orientation between protein and ligand so that the free energy of the overall system is minimized (Gaba et al., 2010).
Plants are widely regarded as reservoir of various types of bioactive molecules with various therapeutic and pharmacological potentials (Raina et al., 2014). Despite the current ascendency of synthesis as a preferred method of drug discovery, the potential of medicinal plants and their compounds to yield new drugs for therapy remains immense (Raskin et al., 2002). Naturally-derived molecules are at the core of the drug discovery process with many more molecules yet to be discovered due to diversity of plant species (Garneau-Tsodikova, 2010). Medicinal plants derived natural products are an immutable source of biologically active agents, they are natural, bio-renewable and readily available unlike synthetic drugs (Ghosh et al., 2008). The Medicinal plant Diospyros mespiliformis has numerous Ethnomedicinal uses including; fevers, pneumonia, syphilis, leprosy and yaws (Von Maydell, 1990).
In this study, three phytochemicals namely; Diospyrin, lupeol and plumbagin isolated by some researchers (Mohamed et al., 2009) were studied in order to determine their pharmacokinetic profile, potential biological activities and toxicity using in silico approaches. The Swiss ADME tool was used to calculate the molecular properties of the ligands following the Lipinski rule of five (5).
MATERIALS AND METHODS
Drug-likeness of the ligands
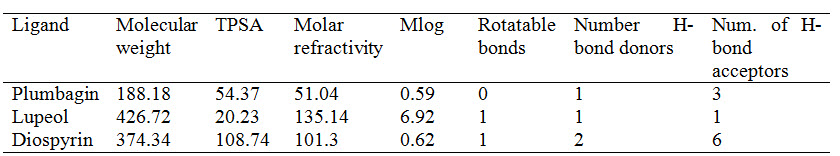
The phytochemicals; diospyrin, plumbagin and lupeol were regarded as ligands. The Swiss ADME tool was used to calculate the molecular properties of the ligands. The molecular properties were screened based on the “Lipinski's rule of five (Lipinski, 2000). The molecular weight, number of rotatable bonds, number of hydrogen bond donors and acceptors, molar refractivity, The total polar surface area (TPSA) and The partition coefficient between n-octanol and water (log Po/w) were calculated using Swiss ADME tool (Daina et al., 2017).
In silico Pharmacokinetic Studies
The chemical structure of the ligands were submitted in the form of canonical simplified molecular input line entry system (SMILE), to estimate several in silico pharmacokinetic parameters (Amina et al., 2016). Using the Swiss ADME tool, the pharmacokinetic profile of the compounds was evaluated. Parameters measured include; human gastrointestinal absorption (HIA) and blood-brain barrier (BBB) permeation both consist in the readout of the BOILED-Egg model (Daina et al., 2016) substrate or non-substrate of the permeability glycoprotein (P-gp), interaction of molecules with cytochromes P450 (CYP) and bioavailability score.
Prediction of Toxicity
Protox-II server (Priyanka et al., 2018) was used to predict the organ toxicities and toxicological end points of the ligands and their LD50. The integrated PubChem search (https:/pubchem.ncbi.nlm.nih.gov/) was used to search for chemical structures using the compound names. The models to be used were selected and the webserver computed the acute toxicity and toxicity targets selected.
Ligand preparation
The ligands were imported from www.zinc15.org in sdf format. Energy minimization was done using Universal force field. The 2D structures of the ligands are shown in Fig I
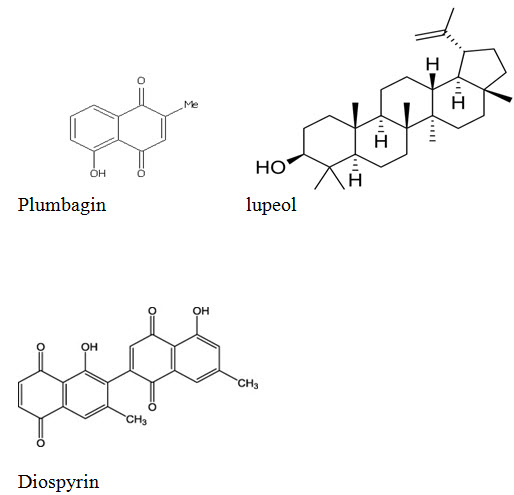
Fig I showing the 2D structures of the ligands
Target Prediction
Swiss target prediction server was used to identify the various potential target based on fit score. The ligands were submitted in the SMILE format. The target set was limited to human targets, and all other parameters were kept as default (David et al., 2014).
Protein preparation
The 3D structures of protein targets identified by Swiss target prediction server were retrieved from the Protein data bank (http://www.rcsb.com) (Gustafson et al., 2014, Patel et al., 2004, Kadavath et al., 2015). The proteins for docking were prepared using the protein preparation wizard of Auto dock. Water molecules present in the crystal structure were removed in the protein preparation process (Morris et al., 2009). Figure II shows the 3D structures of the proteins.
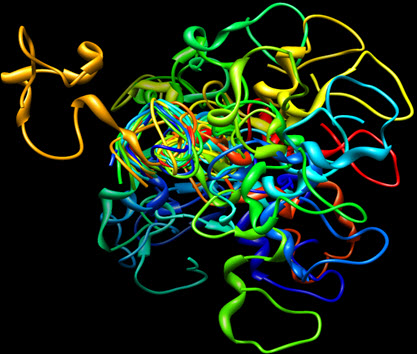
Figure II showing the 3D structure of Microtubule associated protein tau (MAPT)
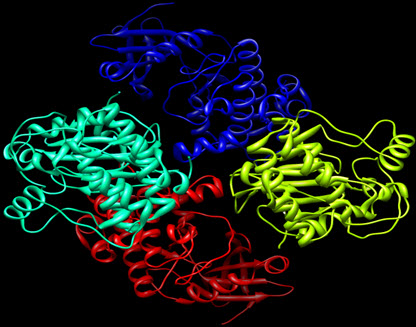
Figure III: showing 3D Structure of Aurora kinase A
In silico molecular Docking Studies
Molecular docking study was conducted using AutoDock vina in The Python Prescription (PyRx) 0.8 virtual screening tool (Trott, 2010). The grid points in X, Y and Z axis were set were set respectively. The grid center was placed in the active site pocket center. Protein and ligands were converted to pdb.qt files. Default docking algorithms were set in conformation with the standard docking protocol. Independent docking runs were carried out for each ligand-protein complex. The results were ranked in the order of increasing docking energies. The lowest binding energy of each cluster was taken as representative (Morris et al., 2009).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESULTS AND DISCUSSIONS
Drug-likeness of the ligands
According to the Lipinski's rule of five, an orally active drug should not violate more than one of the following criteria: lessthan5hydrogen-bonddonors, less than10hydrogen-bondacceptors, a molecular mass less than 500 and log P not greater than 5. All ligands in the present study satisfied the rule. The other significant properties such as total polar surface area (TPSA) and the number of rotatable bonds and molar refractivity were also calculated. The results are depicted in Table 1. TPSA of a compound should be less than 140Å2 and the number of rotatable bonds should be less than 10 (Veber et al., 2002). Plumbagin, lupeol and diospyrin have molecular weight less than 500. Drug molecules with less than 500 are readily transported and have better absorption compared to molecules with higher molecular weight (Srimai et al., 2013).
Table 1 : Molecular Properties of the ligands
In silico Pharmacokinetic Studies
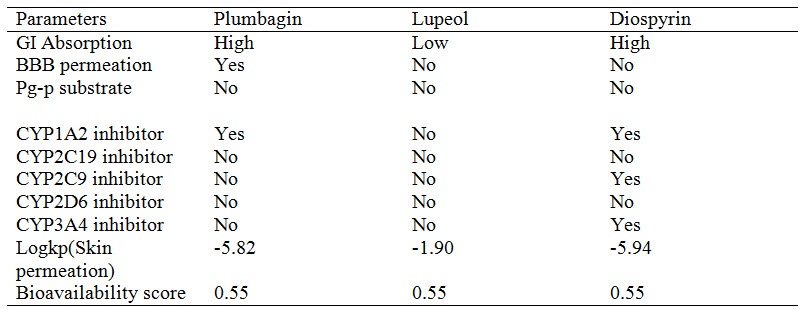
Pharmacokinetic studies such as absorption, distribution and metabolism of the ligands was done using SwissADME (Daina et al., 2016). Blood-Brain Barrier (BBB) penetration, HIA (human Intestinal Absorption), skin permeation, bioavailability score and substrate of the permeability glycoprotein were calculated. The compounds in this study, diospyrin and plumbagin showed high gastrointestinal absorption while lupeol showed low absorption. All the compounds were not substrates for Permeability glycoprotein (P-gp). The information about compounds being substrate or non-substrate of the permeability glycoprotein is key to estimate active efflux through biological membranes, for instance from the gastrointestinal wall to the lumen or from the brain (Montanari, 2015). P-glycoprotein limits cellular uptake of drugs resulting in therapeutic failure because the drug concentration would be lower than expected (Levin, 2012, Lin, 2003). The interaction of molecules with cytochromes P450 isoforms CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 is a significant factor in drug elimination through metabolic biotransformation (Testa 2007). Inhibition of these isoenzymes is an important cause of pharmacokinetics-related drug-drug interactions (Hollenberg, 2002). Some of the cytochrome P450 isoforms could be inhibited by one or more of the compounds. The skin permeability (logP) is a significant parameter for assessment of drugs that might require transdermal administration (Singh, 1993). The more negative the log Kp, the less skin permeate is the molecule. All the compounds in this study are found not to be impermeable through skin. The calculated parameters are presented in Table 2.
Table 2 : Pharmacokinetics
Prediction of Toxicity
The prediction of toxicity was based on 6 different targets linked to adverse drug-reactions. The hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity and cytotoxicity of the compounds were determined (Lounkine et al., 2012). Plumbagin is found to have both mutagenicity and carcinogenicity. Lupeol and diospyrin are reported to be immunotoxic (Table 3). The acute toxicity of the ligands was checked using PROTOX -II server (Priyanka et al., 2018). Lupeol with LD50 of 2000mg/kg has class 4, toxicity. Diospyrin and plumbagin have 16mg/kg and toxicity class 2 respectively (UNECE, 2011).
Table 3 : Toxicity Prediction of Ligands

In silico molecular Docking Studies
The 3D structures of Microtubule assisted protein tau and Aurora kinase A were downloaded from the protein data bank (Gustafson et al., 2014, Patel et al., 2004). The target prediction suggests that plumbagin and lupeol have high preference for Microtubule-associated protein tau (MAPT) with probability of 0.72 and 0.83 respectively. The best target for diospyrin was Aurora kinase A. Plumbagin and lupeol were docked against Microtubule associated protein tau. The dockings scores based on binding energy were; plumbagin -33.8 (kcal/mole) and lupeol -44.7 (kcal/mole). Aurora kinases (Aurora kinase A, B and C) are important and indispensable in multiple steps of mitotic progression (Nigg 2007). Overexpression or amplification of Aurora kinases is generally detected in amount of human cancers, such as breast cancer and is associated with the poor prognosis (Cirak et al., 2015, Ferchichi et al., 2013, Zekri et al., 2012). Aurora kinases become promising therapeutic targets and numerous Aurora kinase inhibitors (AKIs) have been developed (Anqun et al., 2017). In this study, diospyrin showed a binding energy of -10.7 (kcal/mole) and may serve as a potential AKI. The results are shown in Table 4.
Table 4 : In silico Molecular Docking

NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
CONCLUSION
The ligands in this study were found to have good pharmacokinetic profiles. But more studies need to be conducted to ascertain the safety of the compounds. Plumbagin and lupeol showed high affinity for Microtubule associated protein tau. Diospyrin showed affinity for Aurora kinase A and may serve as a potential Aurora kinase A inhibitor (AKI).
REFERENCES
1. Amina I. Dirar, Ayman Y. Waddad, Magdi A. Mohamed, Malik S. Mohamed, Wadah J. Osman, Mona S. Mohammed, Mohamed A. A. Elbadawi (2016) ; In Silico Pharmacokinetics and molecular docking of three leads isolated from Tarconanthus camphoratus L, International Journal of Pharmacy and Pharmaceutical Sciences; 8(5); 975-1491
2. Anqun Tang, Keyu Gao, Laili Chu, Rui Zhang, Jing Yang and Junnian Zheng (2017) Aurora kinases: novel therapy targets in cancers, Oncotarget; 8(14); 23937-23954
3. Cirak Y, Furuncuoglu Y, Yapicier O, Aksu A, Cubukcu E (2015) ; Aurora A overexpression in breast cancer patients induces taxane resistance and results in worse prognosis; J BUON; 20(6); 1414–19.
4. Cross J.B, Thompson D.C, Rai BK, Baber J.C, Fan K.Y, Hu Y, Humblet C (2009) ; Comparison of several molecular docking programs: pose prediction and virtual screening accuracy; J Chem Inf Model; 49(6); 1455–1474
5. Dahlin, J. L., Inglese, J. & Walters, M. A (2015) ; Mitigating risk in academic preclinical drug discovery; Nature Rev. Drug Discov; 14(4); 279–294
6. Daina A., Michielin O., Zoete V (2017) ; SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules ; Sci. Rep; 7; 42717
7. Daina, A., Zoete, V. A (2016) ; BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules; ChemMedChem; 11(11): 1117–1121
8. David Gfeller, Aurélien Grosdidier, Aurélien Grosdidier, Matthias Wirth, Matthias Wirth, Antoine Daina, Olivier Michielin, Vincent Zoete (2014) ; SwissTargetPrediction: a web server for target prediction of bioactive small molecules; Nucleic Acids Research; 42 (Web Server issue):W32-8
9. Ferchichi I, Sassi Hannachi S, Baccar A, Marrakchi Triki R, Cremet J.Y, Ben Romdhane K, Prigent C, Ben Ammar El Gaaied A (2013); Assessment of Aurora A kinase expression in breast cancer: a tool for early diagnosis? ; Dis Markers; 34(2); 63–69.
10. Gaba Monika, Gaba Punam, Singh Sarbjot, and Gupta G. D (2010) An Overview On Molecular Docking, International Journal of Drug Development & Research ; 2(2) ; 219-231
11. Garneau-Tsodikova S (2010); Special series: natural products at the core of drug discovery. Biopolymers; 93(9); 753–4.
12. Ghosh, A., Das, B., Roy, A., Mandal, B. and Chandra, G., (2008); Antibacterial activity of some medicinal plant extracts; Journal of Natural Medicines ; 62(2); 259–262.
13. Gustafson, W.C., Meyerowitz, J.G., Nekritz, E.A., Chen, J., Benes, C., Charron, E., Simonds, E.F., Seeger, R., Matthay, K.K., Hertz, N.T., Eilers, M., Shokat, K.M., Weiss, W.A. (2014) ; Drugging MYCN through an allosteric transition in Aurora Kinase A ; Cancer Cell; 26(3); 414-427.
14. Hollenberg, P. F (2002) ; Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes; Drug Metab.Rev; 334(1-2); 17–35.
15. Kadavath, H., Jaremko, M., Jaremko, M., Biernat, J., Mandelkow, E., Zweckstetter, M. (2015) ; Folding of the Tau Protein on Microtubules; Angew Chem Int Ed Engl. ; 54(35); 10347-10351.
16. Kitchen D.B, Decornez H, Furr J.R, Bajorath J (2004); Docking and scoring in virtual screening for drug discovery: methods and applications. Nature reviews Drug discovery; 3 (11); 935-49
17. Levin G.M (2012); P-glycoprotein: why this drug transporter may be clinically important; Curr Psychiatry; 11(3); 38–40.
18. Lin JH, Yamazaki M (2003); Role of P-glycoprotein in pharmacokinetics: clinical implications; Clin Pharmacokinet; 42 (1); 59-98.
19. Lipinski C.A (2000) ; Drug- like properties and the causes of poor solubility and poor permeability; J.Pharmacol.Toxicol.Methods ; 44(1); 235–249.
20. Lounkine, E., Keiser, M.J., Whitebread, S., Mikhailov, D., Hamon, J., Jenkins, J.L., Lavan, P., Weber, E., Doak, A.K., Cˆot´e, S. et al (2012); Large-scale prediction and testing of drug activity on side-effect targets; Nature; 486(7403); 361–367.
21. Mohamed IE, El Nur EE, Choudhary MI, Khan SN. (2009); Bioactive natural products from two Sudanese medicinal plants Diospyros mespiliformis and Croton zambesicus Rec. Nat. Prod 2009; 3(4); 198-203
22. Montanari, F. & Ecker, G. F (2015) Prediction of drug-ABC-transporter interaction–Recent advances and future challenges; Adv. Drug Deliv. Rev; 86; 17–26.
23. Moroy G, Martiny VY, Vayer P, Villoutreix BO, Miteva MA (2012). Towards in silico structure-based ADMET prediction in drug discovery. Drug Discovery Today; 17: 44–55.
24. Morris, G.M. Huey, R. Lindstrom, W et al (2009) ; AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility; J. Comput Chem; 30(16); 2785-2791.
25. Nigg E.A (2001) ; Mitotic kinases as regulators of cell division and its checkpoints ; Nat Rev Mol Cell Biol; 2(1); 21–32
26. Pakomwit S, Prashant M, Paul T, Cross M, Coster MJ, Gorse AD, Krasavin M, Hofmann A (2015) ; Panel docking of small-molecule libraries: prospects to improve efficiency of lead compound discovery; Biotechnol Adv; 33(6 Pt 1); 941-947.
27. Patel, S., Vuillard, L., Cleasby, A., Murray, C.W., Yon, J.R. (2004); Apo and inhibitor complex structures of BACE (beta-secretase); J Mol Biol; 343(2); 407-16
28. Pelkonen O, Turpeinen M, Raunio H (2011) ; In vivo-in vitro-in silico pharmacokinetic modelling in drug development: current status and future directions; Clin Pharmacokinet; 50(8); 483–491.
29. Priyanka Banerjee, Andreas O Eckert, Anna K Schrey, and Robert Preissner (2018) ; ProTox-II: a webserver for the prediction of toxicity of chemicals, Nucleic Acids Res ; 46(W1); W257–W263.
30. Pugazhendhi .D, Umamaheswari T.S (2013); In silico Methods in Drug Discovery - A Review; International Journal of Advanced Research in Computer Science and Software Engineering; 3(5); 680-683
31. Raina, H., Soni, G., Jauhari, N., Sharma, N., Bharadvaja, N (2014); Phytochemical importance of medicinal plants as potential sources of anticancer agents; Turkish Journal of Botany; 38(6); 1027-1035.
32. Raskin, I., Ribnicky, D., Komarnytsky, S., Ilic, N., Poulev, A., Borisjuk, N., Brinker, A., Moreno, A., Ripoll, C., Yakoby, N., Cornwell, T., Pastor, I. and Fridlender, B (2002) ; Plants and human health in the twenty-first century; Trends In Biotechnology; 20(12); 522-31
33. Singh S, Singh J (1993) ; Transdermal drug delivery by passive diffusion and iontophoresis: a review. Med Res Rev; 13(5); 569–621.
34. Srimai V, Ramesh M, Parameshwar KS, Parthasarathy T (2013); Computer-aided design of selective cytochrome P450 inhibitors and docking studies of alkylresorcinol derivatives; Med Chem Res; 22(11); 5314–23.
35. Testa, B. & Kraemer, S. D (2007); The Biochemistry of Drug Metabolism An Introduction Testa Chemistry & Biodiversity; Wiley Online Library; 4(9); 2031-2122
36. Trott O., Olson A.J (2010) ; AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading; J. Comput. Chem; 31(2); 455-461.
37. UNECE (United Nations Economic Commission for Europe) (2011) Globally Harmonized System of Classification and Labelling of Chemicals (GHS). 4th Rev. New York: United Nations;. http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev04/English/ST-SG-AC10-30-Rev4e.pdf">Rev4e.pdf
38. Van de Waterbeemd H (2002) High throughput and in silico techniques in drug metabolism and pharmacokinetics; Curr. Opin. Drug Disc. Dev; 5 (1); 33–43.
39. Veber D.F., Johnson S.R., Cheng H.Y.et al (2002) Molecular properties that influence the oral bioavailability of drug candidates; J.Med.Chem; 45(12); 2615–2623
40. Von Maydell H (1990); Trees and shrubs of the Sahel : their characteristics and uses; Weikersheim : Verlag Josef Margraf, Scientific Books; Schriftenreihe der Deutschen Gesellschaft für Technische Zusammenarbeit
41. Zekri A, Lesan V, Ghaffari S.H, Tabrizi M.H, Modarressi M.H (2012) ; Gene amplification and overexpression of Aurora-C in breast and prostate cancer cell lines; Oncol Res; 20(5-6); 241-250
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






