
About Authors:
Narasimha Murthy Yedulapurapu*, Babu Rao. Chandu
Donbosco P.G. College of Pharmacy, 5th mile,
Pulladigunta, Kornepadu (V), vatticherukuru (M),
Guntur, Pin code: 522017, Andhra Pradesh.
*Murthyvedulapurapu@gmail.com
ABSTRACT:
Cefixime Trihydrate is an orally active third generation cephalosporin. It has plasma half-life of 3-4hrs; it is active against Gram+ve as well as Gram-ve bacteria. The present investigation involves the enhancement of dissolution rate of cefixime by using various solid dispersion techniques with a view to prolong the drug release in the gastrointestinal tract and consequently into the plasma. The solid dispersions were formulated by using the Croscarmelose sodium as a disintegrant. The solid dispersions were prepared by solvent evaporation, kneading, Physical mixing, Co-grinding techniques. In solvent evaporation technique, Dichloromethane is used as a solvent and in kneading technique water is used as solvent. The prepared solid dispersions were evaluated for in-vitro dissolution studies. Among the 5 formulations F5 sows the good results.
In-vitro dissolution studies were carried out for 60 min using pH 7.2 dissolution media. In-vitro dissolution studies showed that formulations F2, F3, F4, and F5 releases are 15 min, 15 min, 20 min, and 10 min. All the formulations were subjected to analysis by treating the data according to Zero order and first order equations. Formulation F5 follows zero order and first order drug release.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1618
1. INTRODUCTION
The poor solubility and low dissolution rate of poorly water soluble drugs in the aqueous gastro-intestinal fluids often cause insufficient bioavailability rather than the limited permeation through the epitheliaand the formulation of poorly soluble drugs for oral delivery now presents one of the major challenges to formulation scientists in the industries.
The term ‘solubility’ is defined as maximum amount of solute that can be dissolved in a given amount of solvent. Quantitatively it is defined as the concentration of the solute in a saturated solution at a certain temperature. In qualitative terms, solubility may be defined as the spontaneous interaction of two or more substances to form a homogenous molecular dispersion.
1.1 Solid dispersions:
Solid dispersion was introduced in the early 1970s, refers to a group of solid products consisting of at least two different components, generally a hydrophilic matrix and a hydrophobic drug. There are different approaches which can be used for increasing the dissolution of the poorly soluble drugs.
Chiou and Riegelman defined the term solid dispersion as“a dispersion involving the formation of eutectic mixtures of drugs with water soluble carriers by melting of their physical mixtures”; they classified solid dispersions into the following representative types:
* Simple eutectic mixtures,
* solid solutions,
* glass solutions and glass suspensions,
* amorphous precipitations in a crystalline carrier,
* compound or complex formation, and
* Combinations of the previous five types.
[adsense:468x15:2204050025]
While Corrigan (1985) suggested the definition as being a ‘product formed by converting a fluid drug-carrier combination to the solid state’. This strategy includes complete removal of drug crystallinity, and molecular dispersion of the poorly soluble compound in a hydrophilic polymeric carrier. Solid dispersion is a promising approach to improve the dissolution and bioavailability of hydrophobic drugs. The preparation and storage conditions of solid dispersions are crucial since changes may alter the dissolution characteristics of the active ingredients. The development of solid dispersions as a practically viable method to enhance bioavailability of poorly water-soluble drugs overcame the limitations of previous approaches such as salt formation, solubilization by co solvents, and particle size reduction.
When the solid dispersion is exposed to aqueous media, the carrier dissolves and the drug releases as fine colloidal particles. The resulting enhanced surface area produces higher dissolution rate and bioavailability of poorly water-soluble drugs.
The solubility of Cefixime Trihydrate is soluble in methanol but insoluble in water. Cefixime Trihydrate is absorbed orally as 40 – 50% and 50% excreted unchanged in Urine. Its serum half life is 3 – 4 hours. Because of poor solubility of Cefixime Trihydrate it is prepared as sold dispersions by using various techniques like Physical mixing, Co – grinding method , kneading technique and solvent evaporation technique.
Limited in drug absorption results poor bioavailability of drug. In GI Tract the absorption of drug can be limited by the various factors with the most poor aqueous solubility or poor membrane permeability of the drug molecule. When the active ingredient can be delivered as GIT orally, it first dissolved in intestinal fluids before it reach to systemic circulation. Therefore a drug having poor aqueous solubility will typically exhibit in dissolution rate limitation and absorption and a drug with poor membrane permeability will exhibit the permeation rate absorption limited. So that oral bioavailability of drugs can be improved by the enhancing solubility and dissolution rate of poorly water soluble drugs , and another is enhancing the permeability of poor permeable drugs.
1.3 Preparation of solid dispersions:
Solid dispersions can be prepared by the various methods those are deals with the mixing of matrix and a drug, preferably on a molecular level, while the matrix and drug are generally poorly miscible.
During the preparation of solid dispersion techniques, de-mixing and formation of different phases are observed. Phase separations like crystallization or amorphous of drug clusters formation are difficult to control and therefore unwanted. So the phase separation can be minimized by the rapid cooling procedure. In generally phase separation can beprevented by maintaining a low molecular mobility of matrix and drug during preparation. And also, maintain the driving force by keep the mixture at an elevated temperature, there by maintain miscibility for as long as possible1.
1.4 Advantages of solid dispersion:
1.4.1. Particles with reduced particle size
Molecular dispersions, as solid dispersions, represent the last state on particle size reduction, and after carrier dissolution the drug is molecularly dispersed in the dissolution medium. Solid dispersions apply this principle to drug release by creating a mixture of a poorly water soluble drug and highly soluble carriers. A high surface area is formed, resultingin an increased dissolution rate and, consequently,improved bioavailability2.
1.4.2. Particles with improved wettability
A strong contribution to the enhancement of drug solubility is related to the drug wettability improvement verified in solid dispersions. It was observed that even carriers without any surface activity, such as urea improved drug wettability. Carriers with surface activity, such as cholic acid and bile salts. When used, can significantly increase the wettability property of drug. Moreover, carriers can influence the drug dissolution profile by direct dissolution or co-solvent effects3.
1.4.3. Particles with higher porosity
Particles in solid dispersions have been found to have a higher degree of porosity. The increase in porosity also depends on the carrier properties; for instance, solid dispersions containing linear polymers produce larger and more porous particles than those containing reticular polymers and, therefore, result in a higher dissolution rate. The increased porosity of solid dispersion particles also hastens the drug release profile.
1.4.4. Drugs in amorphous state
Poorly water soluble crystalline drugs, when in the amorphous state tend to have higher solubility. The enhancement of drug release can usually be achieved using the drug in itsamorphous state, because no energy is required to break up the crystal lattice during the dissolution process. In solid dispersions, drugs are presented as supersaturated solutions after system dissolution, and it is speculated that, if drugs precipitate, it is as a metastable polymorphic form with higher solubility than the most stable crystal form.
For drugs with low crystal energy (low melting temperature or heat of fusion), the amorphous composition is primarily dictated by the difference in melting temperature between drug and carrier. For drugs with high crystal energy, higher amorphous compositions can be obtained by choosing carriers, which exhibit specific interactions with them.
1.5 Evaluation of prepared solid dispersions Percentage yield
Percentage practical yield were calculated to know about percent yield or efficiency of anyMethod, thus it helps in selection of appropriate method of production. Solid dispersions were collected and weighed to determine practical yield (PY) from the following equation
Practical mass (Solid dispersion)
Percentage yield= ----------------------------------- x 100
Theoretical mass (Drug + carrier)
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
1.5.1 Drug content
The Physical mixture and solid dispersion equivalent to 50 mg of drug were taken and dissolved separately in100 ml of phosphate buffer pH 7.2. The solutions were filtered and were further diluted such that the absorbance falls within the range of standard curve. The absorbances of solutions were determined at 288 nm by UV?visible spectrophotometer. The actual drug content was calculated using the following equation as follows
Practical drug content
% drug content = --------------------------- x 100
Theoretical drug content
1.6 Solid dispersion techniques:
1.6.1 Solvent evaporation method:
Basic process of preparing solid dispersion consists of dissolving the drug and the polymeric carrier in a common solvent such as ethanol, chloroform, or a mixture of ethanol and dichloromethane. In some cases, large volume of solvents as well as heating may be required to enable complete dissolution of drug and carrier. To minimize the volume of organic solvent required, some investigators have reported the use of co-solvents. The main advantage of the solvent method is thermal decomposition of drugs or carriers can be prevented because of the relatively low temperatures required for the evaporation of organic solvents. However, solvent methods show many disadvantages such as; expensive, ecological, and difficult to find common and removable solvents, difficulty in completely removing liquid solvent, difficulty of reproducing crystal form5.
|
Solvent |
Melting point(0c) |
Boiling point(0c) |
Vapour pressure at 25°C (kPa) |
|
Water |
0 |
100 |
3.16 |
|
Methanol |
-93.9 |
65 |
16.9 |
|
Ethanol |
-117 |
78.5 |
5.79 |
|
1-propanol |
-85.8 |
97.4 |
2.27 |
|
2-propanol |
-127 |
82.4 |
5.85 |
|
Chloroform |
-63 |
62 |
26.1 |
|
Dimethylsulphoxide |
19 |
189 |
0.08 |
|
Acetic acid |
17 |
118 |
1.64 |
|
1,4-dioxane |
12 |
102 |
4.92 |
|
2-methyl-2-propanol (TBA) |
25 |
82 |
5.49 |
Table 1.1: Different solvents used
1.6.2 Fusion method/melting method:
The fusion method is sometimes referred to as the melt method.The first solid dispersions created for pharmaceutical applications were prepared by the fusion method. The melting or fusion method was first proposed by Sekiguchi and Obi to prepare fast release solid dispersion dosage forms. The physical mixture of a drug and a water-soluble carrier was heated directly until it gets melted. The melted mixture was then cooled and solidified rapidly in an ice bath under rigorous stirring. The final solid mass was crushed, pulverized, and sieved. Such atechnique was subsequently employed with some modification by Goldberg et al. and Chiou and Riegelman. The solidified masses were often found to require storage of 1 or more days in desiccators at ambient temperatures for hardening and ease of powdering. The fusion method has serious limitations. Firstly, a major disadvantage is that the method can only be applied when drug and matrix are compatible and when they mix well at the heating temperature. When drug and matrix are incompatible two liquid phases or a suspension can be observed in the heated mixture, which results in an inhomogeneous solid dispersion. This can be prevented by using surfactants. Secondly, a problem can arise during cooling when the drug-matrix miscibility changes. In this case phase separation can occur. Indeed, it was observed that when the mixture was slowly cooled, crystalline drug occurred, whereas fast cooling yielded amorphous solid dispersions. Thirdly, degradation of the drug and or matrix can occur during heating to temperatures necessary to fuse matrix and drug6.
1.6.3 Hot melt extrusion:
Hot-melt extrusion (HME) technique represents a novel application of polymer processing technology to prepare pharmaceutical dosage forms. The process involves embedding a drug in a polymer while shaping the composite material to form a pharmaceutical product. This technique is same as the fusion method. The only difference is that in this method, intense mixing of the components is induced by the extruder. High shear forces results in to the high local temperature in the extruder and that can be problematic for the heat sensitive materials. When compared to melting in a vessel, the product stability and dissolution are similar, but melt extrusion offers the potential to shape the heated drug-matrix mixture into implants, ophthalmic inserts, or oral dosage forms. Just like in the traditional fusion process, miscibility of drug and matrix can be a problem. Solubility parameters are investigated to predict the solid state miscibility and to select matrices suitable for melt extrusion. High shear forces resulting in high local temperatures in the extruder are a problem for heat sensitive materials7.
1.6.4 Supercritical fluid technology (SCF):
SCF techniques can be adopted for the preparation of solvent free solid dispersion dosage forms to enhance the solubility of poorly soluble compounds. Super critical fluid is the one where substances existing as a single fluid phase above their critical temperature and pressure. Methodology includes a very fine dispersion of hydrophobic drug in the hydrophilic carrier. Carbon dioxide is the most commonly used SCF because it is chemically inert, non toxic and non flammable8.
1.6.5 Dropping method:
The dropping method was developed by Bulau and Ulrich (1977) to facilitate the crystallization of different chemicals. This method is a new procedure for producing round particles from melted solid dispersions. Methodology includes that the solid dispersion of a melted drug–carrier mixture is dropped onto a cooling plate, where it get solidifies into round particles. The size and shape of the particles can be influenced by factors such as the viscosity of the melt and the size of the pipette. As viscosity is highly temperature dependent, it is very important to adjust the temperature so that, when the melt is dropped onto the plate, it solidifies into a spherical shape. The dropping method does not use organic solvents and therefore has none of the problems associated with solvent evaporation9.
1.6.6 Electrostatic Spinning Method:
This technology is used in polymer industry where in it combines solid solution/dispersion technology with nanotechnology. In this process, a potential between 5 and 30 kV is applied on the liquid stream of a drug/polymer solution. And as when the electrical forces overcome the surface tension of the drug/polymer solution at the air interface, fibers of submicron diameter are formed. After evaporating the solvent, the formed fibers can be collected on a screen10.
1.6.7 Co-precipitation method:
In this method, while during constant stirring, a non solvent is added drop wise to the drug and carrier solution and the drug and carrier are co-precipitated to get micro particles, and then this micro particle suspension is filtered and dried11.
1.7 Characterization of solid dispersion
1.7.1 Detection of crystallinity in solid dispersions:
Several different molecular structures of the drug in the matrix can be encountered in solid dispersions. Many attempts have been made to investigate the molecular arrangement in solid dispersions. However, most effort has been put into differentiate between amorphous and crystalline material. For that purpose many techniques are available which detect the amount of crystalline material in the dispersion. The amount of amorphous material is never measured directly but is mostly derived from the amount of crystalline material in the sample. It should be noted that through the assessment of crystallinity as method to determine the amount of amorphous drug it will not be revealed whether the drug is present as amorphous drug particles or as molecularly dispersed molecules.
1.7.2 Currently, the following techniques are available to detect thedegree of crystallinity
1. Powder X-ray diffraction can be used to qualitatively detect material with long range order. Sharper diffraction peaks indicate more crystalline material. Recently developed X-ray equipment is semi- quantitative.
2. Infrared spectroscopy (IR) can be used to detect the variation in the energy distribution of interactions between drug and matrix. Sharp vibrational bands indicate crystallinity. Fourier Transformed Infrared Spectroscopy (FTIR) was used to accurately detect crystallinities ranging from 1 to 99% in pure material. However in solid dispersions only qualitative detection was possible
3. Water vapour sorption can be used to discriminate between amorphous and crystalline material when the hygroscopicity is different. This method requires accurate data on the hygroscopicity of both completely crystalline and completely amorphous samples.
4. Isothermal Microcalorimetry measures the crystallization energy of amorphous material that is heated above its glass transition temperature. However, this technique has some limitations. Firstly, this technique can only be applied if the physical stability is such that only during the measurement crystallization takes place. Secondly, it has to be assumed that all amorphous material crystallizes. Thirdly, in a binary mixture of two amorphous compounds a distinction between crystallization energies of drug and matrix is difficult.
5. Dissolution Calorimetric measures the energy of dissolution, which is dependent on the crystallinity of the sample. Usually, dissolution of crystalline material is endothermic, whereas dissolution of amorphous material is exothermic.
6. Macroscopic techniques that measure mechanical properties that are different for amorphous and crystalline material can be indicative for the degree of crystallinity. Density measurements and Dynamic Mechanical Analysis (DMA) determine the modulus of elasticity and viscosity and thus affected by the degreeof crystallinity. However, also these techniques require knowledge about the addictively of these properties in intimately mixed binary solids.
7. A frequently used technique to detect the amount of crystalline material is Differential Scanning Calorimetry. In DSC, samples are heated with a constant heating rate and the amount of energy necessary for that is detected. With DSC the temperatures at which thermal events occur can be detected. Thermal events can be a glass to rubber transition, (re)crystallization, melting or degradation. Furthermore, the melting and (re)crystallization energy can be quantified. The melting energy can be used to detect the amount of crystalline material. Possibly, the recrystallization energy can be used to calculate the amount of amorphous material provided, that all amorphous material is transformed to the crystalline state.
1.7.3 Detection of molecular structure in amorphous solid dispersions
The properties of a solid dispersion are highly affected by the uniformity of the distribution of the drug in the matrix. The stability and dissolution behaviour could be different for solid dispersions that do not contain any crystalline drug particles, i.e. solid dispersions of type V and VI or for type II and III. However, not only the Knowledge on the physical state (crystalline or amorphous) is important; the distribution of the drug as amorphous or crystalline particles or as separate drug molecules is relevant to the properties of the solid dispersion too. Nevertheless, only very few studies focus on the discrimination between amorphous incorporated particles versus molecular distribution or homogeneous mixtures.
1. Confocal Raman Spectroscopy was used to measure the homogeneity of the solid mixture of ibuprofen in PVP. It was described that a standard deviation in drug content smaller than 10% was indicative of homogeneous distribution. Because of the pixel size of 2 µm3, uncertainty remains about the presence of nano-sized amorphous drug particles.
2. Using IR or FTIR, the extent of interactions between drug and matrix can be measured. The interactions are indicative for the mode of incorporation of the drug, because separately dispersed drug molecules will have more drug-matrix interactions than when the drug is present in amorphous clusters or other multi-molecule arrangements
3. Temperature Modulated Differential Scanning Calorimetry can be used to assess the degree of mixing of an incorporated drug. Due to the modulation, reversible and irreversible events can be separated. For example, glass transitions (reversible) are separated from crystallization or relaxation (irreversible) in amorphous materials. Furthermore, the value of the Tg is a function of the composition of the homogeneously mixed solid dispersion. It has been shown that the sensitivity of TMDSC is higher than conventional DSC. Therefore this technique can be used to assess the amount of molecularly dispersed drug, and from that the fraction of drug that is dispersed as separate molecules is calculated.
1.8 Alternative strategies
1.8.1 Spraying on sugar beads using a fluidized bed coating system
The approach involves a fluidized bed coating system, wherein a drug-carrier solution is sprayed onto the granular surface of excipients or sugar spheres to produce either granules ready for tableting or drug-coated pellets for encapsulation in one step. The method has been applied for both controlled- and immediate-release solid dispersions.
Itraconazole coated on sugar sphere, is made by layering onto sugar beads a solution of drug and hydroxyl propylmethylcellulose (HPMC) in an organic solvent of dichloromethane and ethanol.
A solid solution of drug in HPMC is produced upon coating (co solvent evaporation) and controlled drying of coated beads in a closed Wurster process. As this thin film dissolves in water or gastric fluid, the molecularly dispersed itraconazole is released at supersaturated concentration. HPMC acts as a stabilizer to inhibit recrystallization of the itraconazole. The supersaturated solutions of itraconazole are sufficiently stable to allow for absorption and distribution12.
1.8.2 Direct capsule filling
Direct filling of hard gelatin capsules with the liquid melt of solid dispersions avoids grinding-induced changes in the crystallinity of the drug. The filling of hard gelatin capsules has been feasible in molten dispersions of Triamterene-PEG 500 using a Zanasi LZ 64 capsule- filling machine . However, PEG was not a suitable carrier for the direct capsule-filling method as the water-soluble carrier dissolved more rapidly than the drug, resulting in drug-rich layers formed over the surface of dissolving plugs, which prevented further dissolution of the drug. A surfactant must be mixed with the carrier to avoid formation of a drug-rich surface layer (e.g., polysorbate 80 with PEG, phosphatidylcholine with PEG). The temperature of the molten solution should not exceed ~70oC because it might compromise the hard-gelatin capsule shell.
1.8.3 Surface-active carriers
The surface-active and self-emulsifying carriers for solid dispersion of poorly water-soluble drugs have been of great interest in recent years. A surface-active carrier may be preferable in almost all cases for the solid dispersion of poorly water-soluble drugs. Two of the important surface-active carriers are Gelucire 44/14 and Vitamin E R-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS). Gelucire 44/14 has commonly been used in solid dispersion for the bioavailability enhancement of drugs. Gelucire 44/14 is a mixture of glyceryl and PEG 1500 esters of long-chain fatty acids and is official in the European Pharmacopoeia as lauryl macrogolglycerides; the suffixes 44 and 14 in its name refer, respectively, to its melting point and hydrophilic- lipophilic balance (HLB) value. Vitamin E TPGS National Formulary (NF) (Eastman, Kingsport, TN) is prepared by the esterification of the acid group of d-R- tocopheryl acid succinate by PEG 1000. The material has an HLB value of 13 and is miscible with water in all parts. Its melting point, however, is relatively low (38oC), and it may require mixing with other carriers to increase melting temperatures of formulations9.
A commonly used surfactant, Polysorbate 80, when mixed with solid PEG, has also been reported to be an alternative surface-active carrier. Polysorbate 80 is liquid at room temperature; it forms a solid matrix when it is mixed with a PEG because it incorporates within the amorphous regions of PEG solid structure. As much as 75% (wt/wt) Polysorbate80 was incorporated, PEG remained semisolid, and the lowering of the melting temperature of the PEG used was <12oc The PEG-polysorbate carriers have been found to enhance dissolution36 and bioavailability of drugs from the solid dispersions. Incorporation of 5% (wt/wt) phosphatidyl- choline resulted in enhanced dissolution rate of nifedipine from a PEG-based solid dispersion. Pulverized solid dispersions in PEG containing varying amounts of ionic and nonionic surfactants, including sodium dodecyl sulfate and Polysorbate 80 gave increased dissolution rate of drug13.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
2. LITERATURE SURVEY
2.1 DRUG PROFILE14, 15
Cefixime Trihydrate:
It is a third generation cephalosporin antibiotic used in the management of various infections caused by Gram +ve as well as Gram - Ve Bacteria’s.
2.1.1 DESCRIPTION:
Compound: Cefixime Trihydrate
Chemical name: (6R, 7R)-7-[[(Z)-2-(2-aminothiazol-4-yl)-2-
[(carboxymethoxy) imino] acetyl] amino]-
3-ethenyl-8-oxo-5-thia-1-azabicyclo [4.2.0] Oct-
2-ENE-2- Carboxylic Acid trihydrate.
Molecular Formula: C16H15N507S2.3H20
Molecular Weight: 507.50 gm/mole
Description: Cefixime is a white to light yellow powder.
Solubility: Soluble in Methanol, insoluble in water.
Storage: Stored in air tight container protected from light.
Structural Formula:
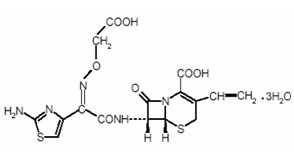
Fig 2.1: Chemical Name of Cefixime Trihydrate
Category: Third generation cephalosporin’s.
Dosage:
Adult The recommended dose is 400mg.
Children The recommended dose is 8mg/kg/day.
(These may be administered as a single daily Dose or may be given in two divided dose)
Table 2.1 Serum Levels of Cefixime in Adults after Administration of Tablets (μg/mL)
|
DOSE |
1hr |
2hr |
4hr |
6hr |
8hr |
12hr |
24hr |
|
100 mg* |
0.3 |
0.8 |
1.0 |
0.7 |
0.4 |
0.2 |
0.2 |
|
200 mg |
0.7 |
1.4 |
2.0 |
1.5 |
1.0 |
0.4 |
0.03 |
|
400 mg |
1.2 |
2.5 |
3.5 |
2.7 |
1.7 |
0.6 |
0.04 |
Table 2.2 Serum Levels of Cefixime in Adults after Administration of Oral Suspension (μg/mL)
|
DOSE |
1hr |
2hr |
4hr |
6hr |
8hr |
12hr |
24hr |
|
100 mg |
0.7 |
1.1 |
1.3 |
0.9 |
0.6 |
0.2 |
0.2 |
|
200 mg |
1.2 |
2.1 |
2.8 |
2.0 |
1.3 |
0.5 |
0.07 |
|
400 mg |
1.8 |
3.3 |
4.4 |
3.3 |
2.2 |
0.8 |
0.07 |
The serum half-life of Cefixime in healthy subjects is independent of dosage form and average 3 to 4 hours but may range up to 9 hours in some normal volunteers.
2.1.2 PHARMACOLOGY:
In rats, 14C-labelled Cefixime was distributed (in order of descending amounts) to the kidneys, lungs, liver, heart, spleen, and brain at 1 hour following a single oral dose of Cefixime and to the kidneys, urinary bladder, blood, liver, and lungs at 5 minutes after a single intravenous dose. In dogs, tissue radioactivity was noted in bile, kidney, liver, lung, testes, heart, and brain after single or multiple intravenous dosing with 14C-labelled Cefixime. After multiple oral dosing, accumulation of Cefixime was negligible in the serum and urine of adult rats and dogs. The doses used in these studies were 100 and 1000 mg/kg/day administered for 1 month to rats and up to 400 mg/kg/day (100, 200 and 400 mg/kg/day) for 53 weeks to dogs.
In addition, there was no evidence of drug accumulation in serum or urine after two weeks of intravenous dosing (320 and 1000 mg/kg/day) in adult dogs. In animal studies, it was noted that Cefixime is excreted in the bile in excess of 10% of the administered dose.
2.1.3 PHARMACOKINETIC PARAMETERES:
Absorption:
Cefixime Trihydrate given orally is about 40%-50% absorbed whether administered with or without food; however, time to maximal absorption is increased approximately 0.8 hours when administered with food.
The oral suspension produces average peak concentrations approximately 25%-50% higher than the tablets. Two hundred milligram doses of oral suspension produce average peak concentrations of 3mcg?ml (range 1 to 4.5 mcg?ml) and 4.6 mcg/ml (range 1.9 to 7.7 mcg/ml), respectively. The area under the time versus concentration curve is greater by approximately 10%-25% with the oral suspension than with the tablet after doses of 100 to 400mg. this increased absorption should be taken into consideration if the oral suspension is to be substituted for the tablet. In children pharmacokinetic of Cefixime Trihydrate are8mg/kg/day.
Distribution:
Cefixime Trihydrate widely distributed I body tissues and fluids. It achieves high concentration in tonsils, maxillary sinus, bile and bile duct. Cefixime Trihydrate about 67% bound to plasma proteins.
Elimination:
No biologically active metabolites of Cefixime Trihydrate have been identified in plasma or urine. Approximately 50% of the absorbed dose is excreted unchanged in urine in 24 hours. The serum half-life of Cefixime Trihydrate in healthy subjects is independent of the dosage forms and averages 3.0-4.0 hours.
Indications:
Cefixime Trihydrate is indicated in the treatment of the following infections when caused by susceptible strains of the designated microorganisms.
* Uncomplicated Urinary Tract infections caused by Escherichia coli and Proteus mirabilis.
* Otitis media caused by Haemophilus influenza (Beta-lactamase positive and negative strains), Maraxella (Branhamella) catarrhalis, and S.pyogenes.
* Pharyngitis and Tonsillitis caused by S.pyogenes.
* Uncomplicated Gonorrhea caused by Neisseria.
2.1.4 Drug interaction:
Carbamazepine: Elevated carbamazepine levels have been reported in post marketing experience when Cefixime Trihydrate administered concomitantly. Drug monitoring may be of assistance in detecting alterations in carbamazepine plasma concentrations.
Warfarin and Anticoagulants: Increased prothrombin time, with or without clinical bleeding, has been reported when Cefixime Trihydrate is administered concomitantly.
2.1.5 Adverse Reactions:
The following adverse reactions have been reported following the use of Cefixime Trihydrate. Incidence rates were less than 1 in 50 (less than 2%).
Gastrointestinal: Diarrhea, loose stools, abdominal pain, dyspepsia, nausea and vomiting. Several cases of documented pseudo membranous colitis symptoms may occur during or after therapy.
Hypersensitivity Reactions: Skin rashes, urticaria, Drug fever and pruritus.
Central nervous system: Headache, Dizziness.
Others: Genital Pruritus, Vaginitis.
2.1.6 Over dosage:
Gastric lavage may be indicated, otherwise, no specific antidote exists. Cefixime Trihydrate is not removed in significant quantities from the circulation by hemodialysis or peritoneal dialysis, Adverse reactions in small numbers of healthy adult volunteers receiving single dose up to 2gm of Cefixime Trihydrate does not differ from the profile seen in patients the recommended doses.
2.1.7 Contraindications:
* Cefixime Trihydrate is contraindicated in patients with known allergy to the cephalosporin group of antibiotics.
* Cefixime Trihydrate should be administered cautiously to any patient who has demonstrated some form of allergy, particularly to antibiotics.
* Treatment with Cefixime Trihydrate alters the normal flora of the colon and many permit overgrowth of clostridia. It is reported that toxin produced by clostridium difficlie is a primary cause of severe antibiotic- associated diarrhea including Pseudo membranous colitis.
2.1.8 Precautions:
Nursing Mothers: It is not known whether Cefixime Trihydrate is excreted in human milk. Consideration should be given to discontinuing temporarily during treatment with this drug.
* Pediatric Use: Safety and effectiveness of Cefixime Trihydrate in Children aged less than six months old have not been established.
2.2 EXCIPIENT PROFILE:
Substances, other than the active ingredient, which have been appropriately evaluated for safety and are included in a drug delivery system to provide support.
The excipients used must have following characteristics-
* They must be stable both physically, chemically and must be biologically inactive.
* It must be free from microbial contamination.
* Excipients used in tablet formulation must be acceptedby regulatory agencies and should meet the entire current regulatory requirement.
2.2.1 CROSCARMELOSE SODIUM
Synonyms: Ac- di sol, Primellose, Vivasol, Solutab, Explocel, Nymcel ZSX.
Empirical formul








