

 About Authors:
About Authors:
Divya Rawat, U.K.Singh, Faizi Muzaffar
Kharvel Subharti College of Pharmacy,
swami Vivekanand subharti university, Subhartipuram,
N.H-58, Meerut By Pass Road, Meerut, Uttar Pardesh-250001, India
*drawat05@gmail.com
Abstract
Microspheres constitute an important part of novel drug delivery system by virtue of their small size and efficient carrier capacity. Microspheres are characteristically free flowing powders consisting of proteins or synthetic polymers having a particle size ranging from 1-1000 μm. The range of Techniques for the preparation of microspheres offers a Variety of opportunities to control aspects of drug administration and enhance the therapeutic efficacy of a given drug. There are various approaches in delivering a therapeutic substance to the target site in a sustained controlled release fashion. One such approach is using microspheres as carriers for drugs also known as micro particles. It is the reliable means to deliver the drug to the target site with specificity, if modified, and to maintain the desired concentration at the site of interest. Microspheres received much attention not only for prolonged release, but also for targeting of anticancer drugs. The purpose of the review is to compile various types of microspheres, different methods to preparation, its applications and also various parameters to evaluate their efficiency.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1713
IMPORTANT IN THIS ARTICLE:
* POLYMERS USED IN MICROSPHERES & METHOD OF PREPARATION
INTRODUCTION
In contrast to drug delivery system, the word novel is searching something out of necessity. The drug has to be delivered for a prolonged period of time and many medicines have to be taken simultaneously in case of chronic patients. Frequent administration of drug is necessary when those have shorter half life and all these leads to decrease in patient’s compliance.[1]
In order to overcome the above problems, various types of controlled release dosage forms are formulated and altered, so that patient compliance increase through prolonged effect , adverse effect decreases by lowering peak plasma concentration.2 The controlled release dosage form maintaining relatively constant drug level in the plasma by releasing the drug at a predetermined rate for an extended period of time. One such in Microspheres as carriers of drug become an approach of controlled release dosage form in novel drug delivery system.[2]
Microspheres are small spherical particles, with diameters in the micrometer range (typically 1 μm to 1000 μm). Microspheres are sometimes referred to as microparticles.
Microspheres can be manufactured from various natural and synthetic materials. Glass microspheres, polymer microspheres and ceramic microspheres are commercially available. Solid and hollow microspheres vary widely in density and, therefore, are used for different applications. Hollow microspheres are typically used as additives to lower the density of a material. Solid microspheres have numerous applications depending on what material they are constructed of and what size they are.
Polyethylene and polystyrene microspheres are two most common types of polymer microspheres. Polystyrene microspheres are typically used in biomedical applications due to their ability to facilitate procedures such as cell sorting and immune precipitation. Proteins and ligands adsorb onto polystyrene readily and permanently, which makes polystyrene microspheres suitable for medical research and biological laboratory experiments. Polyethylene microspheres are commonly used as permanent or temporary filler. Lower melting temperature enables polyethylene microspheres to create porous structures in ceramics and other materials. High sphericity of polyethylene microspheres, as well as availability of colored and fluorescent microspheres, makes them highly desirable for flow visualization and fluid flow analysis, microscopy techniques, health sciences, process troubleshooting and numerous research applications. Charged polyethylene microspheres are also used in electronic paper digital displays.
Glass microspheres are primarily used as filler for weight reduction, retro-reflector for highway safety, additive for cosmetics and adhesives, with limited applications in medical technology. Ceramic microspheres are used primarily as grinding media. Microspheres vary widely in quality, sphericity, uniformity of particle and particle size distribution. The appropriate microsphere needs to be chosen for each unique application.
The term microcapsule is defined as a spherical particle with size varying from 50nm to 2nm containing a core substance. Alternate terminology for the microsphere is micro beads and -beads are used alternatively.[3] [4]
ADVANTAGES
1. Microspheres provide constant and prolonged therapeutic effect.
2. Reduces the dosing frequency and thereby improve the patient compliance.
3. They could be injected into the body due to the spherical shape and smaller size.
4. Better drug utilization will improve the bioavailability and reduce the incidence or intensity of adverse effects.
5. Microsphere morphology allows a controllable variability in degradation and drug release.[5]
LIMITATION
Some of the disadvantages were found to be as follows
1. The modified release from the formulations.
2. The release rate of the controlled release dosage form may vary from a variety of factors like food and the rate of transit though gut.
3. Differences in the release rate from one dose to another.
4. Controlled release formulations generally contain a higher drug load and thus any loss of integrity of the release characteristics of the dosage form may lead to potential toxicity.
5. Dosage forms of this kind should not becrushed or chewed.[5]
Uses:
1. Taste and odour masking.
2. Conversion of oil and other liquids, facilitating ease of handling.
3. Protection of the drugs from the environment.
4. Delay of volatilization.
5. Freedom from incompatibilities between drugs and excipients, especially the buffers.
6. Improvement of flow properties.
7. Safe handling of toxic substances.
8. Dispersion of water insoluble substances on aqueous media.
9. Production of sustained release, controlled release and targeted medications.
10. Reduced dose dumping potential compared to large implantable devices.
11. They facilitate accurate delivery of small quantities of potent drugs and reduced concentration of the drug at sites other than the target organ of tissue.
TYPES OF MICROSPHERES
[adsense:468x15:2204050025]
1. Bio adhesive microspheres:[7]
Adhesion can be defined as sticking of drug to the membrane by using the sticking property of the water soluble polymers. Adhesion of drug delivery device to the mucosal membrane such as buccal, ocular, rectal, nasal etc can be termed as bioadhesion. The term “bioadhesion”describes materials that bind to biological substrates’, such as mucosal members. Adhesion of Bioadhesive drug delivery devices to the mucosal tissue offers the possibility of creating an intimate and prolonged contact at the site of administration. This prolonged residence time can result in enhanced absorption and in combination with a controlled release of drug also improved patient compliance by reducing the frequency of administration. Carrier technology offers an intelligent approach for drug delivery by coupling the drug to a carrier particle such as microspheres, nanospheres, liposomes, nanoparticles, etc., which modulates the release and absorption of the drug. Microspheres constitute an important part of these particulate drug delivery systems by virtue of their small size and efficient carrier capacity.
2. Magnetic microspheres:[8],[9],[10],[11].
This kind of delivery system is very much important which localises the drug to the disease site. In this larger amount of freely circulating drug can be replaced by smaller amount of magnetically targeted drug. Magnetic carriers receive magnetic responses to a magnetic field from incorporated materials that are used for magnetic microspheres are chitosan, dextran etc.[8] The different type are Therapeutic magnetic microspheres are used to deliver chemotherapeutic agent to liver tumour. Drugs like proteins and peptides can also be targeted through this system.[9] Diagnostic microspheres.[10] Magnetic drug transport technique is based on the fact that the drug can be either encapsulated into a magnetic microsphere or conjugated on the surface of the microsphere.The accumulation of the carrier at the target site allow them to deliver the drug locally.[11]
3. Floating microspheres:[12].
In floating types the bulk density is less than the gastric fluid and so remains buoyant in stomach without affecting gastric emptying rate. The drug is released slowly at the desired rate, if the system is floating on gastric content, increases gastric residence and fluctuation in plasma concentration. It also reduces chances of striking and dose dumping and produces prolonged therapeutic effect. Drug (ketoprofen) given through this form.[12]
4. Radioactive microspheres:[13]
Radio emobilisation therapy microspheres sized 10-30 nm are of larger than capillaries and gets tapped in first capillary bed when they come across. They are injected to the arteries that lead to tumour of interest. So these radioactive microspheres deliver high radiation dose to the targeted areas without damaging the normal surrounding tissues. It differs from drug delivery system, as radio activity is not released from microspheres but acts from within a radioisotope typical distance and the different kinds of radioactive microspheres are α emitters, β emitters, γ emitters.[13]
5. Muco adhesive microspheres:[14]
Muco adhesive microspheres which are of 1-1000mm in diameter and consisting either entirely of a muco adhesive polymer or having an outer coating of it and coupling of muco adhesive properties to microspheres has additional advantages, e.g. efficient absorption and enhanced bioavailability of the drugs due to a high surface to volume ratio, a much more intimate contact with the mucus layer, specific targeting of drug to the absorption site achieved by anchoring plant lectins, bacterial adhesions and antibodies, etc. on the surface of the microspheres. Mucoadhesive microspheres can be tailored to adhere to any mucosal tissue including those found in eye, nasal cavity, urinary and gastrointestinal tract, thus offering the possibilities of localized as well as systemic controlled release of drugs.[14]
6. Polymeric microspheres:[15]
The different types of polymeric microspheres can be classified as
(a) Biodegradable polymeric microspheres:
Natural polymers such as starch are used with the concept that they are biodegradable, biocompatible, and also Bioadhesive in nature. Biodegradable polymers prolongs the residence time when contact with mucous membrane due to its high degree of swelling property with aqueous medium , results gel formation. The rate and extent of drug release is controlled by concentration of polymer and the release pattern in a sustained manner. The main drawback is, in clinical use drug loading efficiency of biodegradable microspheres is complex and is difficult to control the drug release.
(b) Synthetic polymeric microspheres:
The interest of synthetic polymeric microspheres are widely used in clinical application, moreover that also used as bulking agent, fillers, embolic particles, drug delivery vehicles etc and proved to be safe and biocompatible. But the main disadvantage of these kind of microspheres, are tend to migrate away from injection site and lead to potential risk, embolism and further organ damage.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
POLYMERS USED IN MICROSPHERES:[16]
Polymers used in the microsphere are generally classified into two types:
1. Synthetic polymers
2. Natural polymers
Table (1): Classification of Polymer:
|
POLYMER |
SUB TYPES |
EXAMPLES |
|
Synthetic polymer |
Biodegradable |
Lactides, Glycolides & their co polymers Poly alkyl cyano acrylates Poly anhydrides |
|
Non–biodegradable |
Poly methyl methacrylate Acrolein Glycidyl methacrylate Epoxy polymers |
|
|
Natural polymer |
Proteins |
Albumin Gelatin Collagen |
|
Carbohydrates |
Agarose Carrageenan Chitosan Starch |
|
|
Chemically modified carbohydrates |
Poly dextran, Poly starch |
Table (2): Various types of polymers and their application:[17],[18],[19]
|
Polymer |
Mechanism |
|
Modified starch, HPMC, Carbopol 974P |
Slower release of drug. |
|
Ethyl Cellulose |
Controlled release for longer period of time |
|
PLGA, Chitosan |
Vaccine delivery |
|
Chitosan coated PLGA microspheres |
Targeted drug delivery |
|
Polyvinylalcohol, Polyacrylamide |
Adsorption of harmful substances in blood |
METHOD OF PREPARATION:
Incorporation of solid, liquid or gases into one or more polymeric coatings can be done by microencapsulation technique. The different methods used for various microspheres preparation depends on particle size, route of administration, duration of drug release, method of cross linking, evaporation time and co-precipitation, etc. The various methods of preparations are:
A. Emulsion Solvent Evaporation Technique.
B. Emulsion Cross Linking Technique.
C. Emulsion-Solvent Diffusion Technique.
D. Emulsification Heat Stabilizing Technique.
E. Co-acervation Phase Separation Technique.
a) Thermal Change.
b) Non-Solvent Addition.
c) Polymer Addition.
d) Salt Addition.
e) Polymer-Polymer Interaction.
F. Spray Drying Technique.
G. Polymerization Technique.
a) Normal polymerization.
b) Interfacial polymerization.
H. Ionic Gelation Technique.
I. Hydroxyl Appetite (HAP) Microspheres In Sphere Morphology.
J. Hot Melt Micro encapsulation technique.
Preparation of microspheres should satisfy certain criteria:
1. The ability to incorporate reasonably high concentrations of the drug.
2. Stability of the preparation after synthesis with a clinically acceptable shelf life.
3. Controlled particle size and dispersibility in aqueous vehicles for injection.
4. Release of active reagent with a good control over a wide time scale.
5. Biocompatibility with a controllable biodegradability and Susceptibility to chemical modification.
A. Emulsion solvent evaporation technique:[20]
In this technique the drug is dissolved in polymer which was previously dissolved in chloroform and the resulting solution is added to aqueous phase containing 0.2 % sodium of PVP as emulsifying agent. The above mixture was agitated at 500 rpm then the drug and polymer (eudragit) was transformed into fine droplet which solidified into rigid microspheres by solvent evaporation and then collected by filtration and washed with demineralised water and desiccated at room temperature for 24 hrs. Aceclofenac microspheres were prepared by this technique.

Solvent evaporation method for preparation of microsphere.
B. Emulsion cross linking method:[21]
In this method drug was dissolved in aqueous gelation solution which was previously heated for 1 hr at 40oC. The solution was added drop wise to liquid paraffin while stirring the mixture at 1500 rpm for 10 min at 35oC, results in w/o emulsion then further stirring is done for 10 min at 15oC. Thus the produced microspheres were washed respectively three times with acetone and isopropyl alcohol which then air dried and dispersed in 5mL of aqueous glutaraldehyde saturated toluene solution at room temperature for 3 hrs for cross linking and then was treated with 100mL of 10mm glyciene solution containing 0.1%w/v of tween 80 at 37oC for 10 min to block un reacted glutaraldehyde.Examples for this technique is Gelatin A microspheres.
C. Emulsion-Solvent Diffusion Technique:[22]
In order to improve the residence time in colon floating microparticles of drug is prepared by emulsion solvent diffusion technique. The drug polymer mixture is dissolved in a mixture of ethanol and dichloromethane (1:1) then the mixture is added drop wise to sodium lauryl sulphate (SLS) solution. The solution is stirred with propeller type agitator at room temperature at 150 rpm for 1 hr, washed and dried in a desiccator at room temperature.
D. Emulsification Heat Stabilizing Technique:[23]
In this method, drug and polymer are dissolved in 20 ml of deionised water and 5 ml of egg albumin solution and 0.1% of Tween?80 are added stirred it for 30 min. The prepared solution is used as aqueous phase. The oil phase is prepared by mixing 20 ml of sunflower oil and 5ml of diethyl ether with 1% span?80 (as emulsifier) and stirred it for 20 mins at 800?1000 rpm on a magnetic stirrer. The primary emulsion is prepared by adding the oil phase drop wise to the aqueous phase followed by stirring it for 30 mins at 800?1000 rpm. The prepared primary emulsion is added to pre?heated (65 to 70?C) sunflower oil (80 ml) by using 21 No. needle and stirred at 1000?1200 rpm for 2 hrs till the solidification of microspheres takes place. The suspension then allowed to cool to room temperature with continuous stirring using a magnetic stirrer. On cooling, 100 ml of anhydrous ether is added. The suspension containing the microspheres is centrifuged for 15 mins and the settled microspheres are washed three times with ether to remove traces of oil on microspheres surfaces. The obtained microspheres are then vacuum dried in a desiccator overnight and stored at 4?C in dark.
E. Co-acervation Phase Separation Technique:[24]
a) Thermal Change: Microspheres are formed by dissolving polymer (ethyl cellulose) in cyclohexane with vigorous stirring at 80 ?C by heating. Then the drug is finely pulverized and added to the above solution with vigorous stirring. The phase separation is brought about by reducing temperature using ice bath. The product is washed twice withcyclohexane and air dried then passed through sieve (sieve no. 40) to obtain individual microcapsule.
b) Non Solvent Addition: Microspheres are formed by dissolving polymer (ethyl cellulose) in toluene containing propyl-isobutylene in a closed beaker with stirring for 6 hrs. at 500 rpm and the drug is dispersed in it. Stirring is continued for 15 mins., then phase separation is brought about by petroleum benzene with continuous stirring. The microcapsules washed with n-hexane and air dried for 2 hrs., and kept in an oven at 50?C for 4 hrs
c) Polymer Addition: Microspheres are formed by dissolving polymer (ethyl cellulose) is dissolved in toluene, then1 part is added to 4 parts of crystalline methylene blue hydrochloride. Co-acervation is accomplished by adding liquid polybuta-diene. Then the polymer coating is solidified by adding a nonsolvent (hexane). The resulting product is washed and air dried.
d) Salt Addition: Microspheres are formed by dissolving oil soluble vitamin in corn oil and is emulsified by using pig skin gelatin under condition of temperature 50?C, coacervation is induced by adding sodium sulphate. Stirring is necessary for uniform coating of gelatin. The resultant microspheres product is collected and washed with water, chilled below gelation temperature of gelatin and dried by using spray drying.
e) Polymer-Polymer Interaction: In this process, aqueous solution of gum Arabica and gelatin (isoelectic point 8.9) are prepared, the homogeneous polymer solutions are mixed together in equal amount, diluted to about twice their volume with water, adjusted to pH 4.5 and warmed to 40- 45?C. the oppositely charged macromolecules interact at these conditions and undergo co-acervation. While maintaining the warm temperature, the liquid core material (methyl salicylate) is added to polmer solution and stirred well. Then the mixture is cooled to 25?C and coating is rigidised by cooling the mixture to 10?C.
F. Spray drying technique:[25]
This was used to prepare polymeric blended microsphere loaded with ketoprofen drug. It involves dispersing the core material into liquefied coating material and then spraying the mixture in the environment for solidification of coating followed by rapid evaporation of solvent.4 Organic solution of poly (epsilon-caprolactone) (PCL) and cellulose acetate butyrate (CAB), in different weight ratios and ketoprofen were prepared and sprayed in different experimental condition achieving drug loaded microspheres. This is rapidbut may loose crystalinity due to fast drying process.
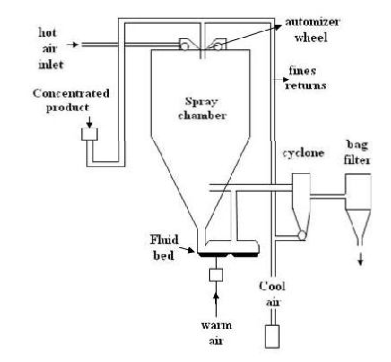
Spray drying method for preparation of microsphere.
G. Polymerization Techniques:[26],[27].
Mainly two techniques are used for the preparation of microsphere by polymerization technique:
(a) Normal polymerization:
Normal polymerization classified as:
1. Bulk polymerization
2. Suspension/ pearl polymerization
3. Emulsion polymerization
1. In bulk polymerization, a monomer or a mixture of monomers along with the initiator or catalyst is usually heated to initiate polymerization. Polymer obtained may be moulded as microspheres. Drug loading may be done by adding the drug during the process of polymerization. It is a pure polymer formation technique but it is very difficult to dissipate the heat of reaction which affects the thermo labile active ingredients.
2. Suspension polymerization is carried out at lower temperature and also referred to as pearl polymerization in which the monomer mixture is heated with active drug as droplets dispersion in continuous aqueous phase. Microsphere size obtained by suspension techniques is less the 100 μm.
3. Emulsion polymerization differs from the suspension polymerization due to presence of initiator in aqueous phase and also carried out at low temperature as suspension. External phase normally water in last two techniques so through which heat can be easily dissipated. The formation of higher polymer at faster rate is possible by these techniques but sometimes association of polymer with the un- reacted monomer and other additives can occur.
(b) Interfacial polymerization:
It involves the reaction of various monomers at the interface between the two immiscible liquid phases to form a film of polymer that essentially envelops the dispersed phase. In this technique two reacting monomers are employed; one is dissolved in continuous phase while other is dispersed in continuous phase (aqueous in nature) throughout which the second monomer is emulsified. Two conditions arise because of the solubility of formed polymer in the emulsion droplet. The formation is Monolithic, if the polymer is soluble in droplet and the formation is Capsular type if the polymer is insoluble in droplet.
H. Ionic gelation:[28]
Alginate/chitosan particulate system for diclofenac sodium release was prepared using this technique. 25 % (w/v) of diclofenac sodium was added to 1.2 % (w/v) aqueous solution of sodium alginate. In order to get the complete solution stirring is continued and after that it was added dropwise to a solution containing Ca2+ /Al3+ and chitosan solution in acetic acid. Microspheres which were formed were kept in original solution for 24 hr for internal gellification followed by filteration for separation. The complete release was obtained at pH 6.4-7.2 but the drug did not release in acidic pH.
I. Hydroxyl appetite (HAP) microspheres in sphere morphology:[28]
This was used to prepare microspheres with peculiar spheres in sphere morphology microspheres were prepared by o/w emulsion followed by solvent evaporation. At first o/w emulsion was prepared by dispersing the organic phase (Diclofenac sodium containing 5% w/w of EVA and appropriate amount of HAP) in aqueous phase of surfactant. The organic phase was dispersed in the form of tiny droplets which were surrounded by surfactant molecules this prevented the droplets from co solvencing and helped them to stay individual droplets .While stirring the DCM was slowly evaporated and the droplets solidify individual to become microspheres.
J. Hot Melt Microencapsulation Technique:[29]
The polymer is first melted and then mixed with solid particles of the drug that has been sieved to less than 50 μm. The mixture is suspended in a non-miscible solvent (like silicone oil), continuously stirred, and heated to 5°C above the melting point of the polymer. Once the emulsion is stabilized, it is cooled until the polymer particles solidify. The resulting microspheres are washed by decantation with petroleum ether. The primary objective for developing this method is to develop a microencapsulation process suitable for the water labile polymers, e.g. polyanhydrides. Microspheres with diameter of 1-1000 μm can be obtained and the size distribution can be easily controlled by altering the stirring rate. The only disadvantage of this method is moderate temperature to which the drug is exposed.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
CHARECTERIZATION/ EVALUATION OF MICROSPHERES
1. Interaction study by TLC/ FTIR.[30]
IR spectroscopic studies:-The IR spectra of the free drug and the microspheres are recorded. The identical peaks corresponding to the functional groups features confirm that neither the polymer nor the method of preparation has affected the drug stability.
Thin layer chromatographic studies:-The drug stability in the prepared microspheres canalso be tested by the TLC method. The Rf values ofthe prepared microspheres can be compared with theRf value of the pure drug. The values indicate thedrug stability.
UV-FTTR (Fourier transform infra red):-The drug polymer interaction and also degradation of drug while processing for microencapsulation can be determined by FTIR. In this method the pellets of drug and potassium bromide are prepared by compressing the powders at 20 psi for 10 min on KBr?press and the spectra are scanned in the wave number range of 4000?600 cm?1. FTIR study is carried on pure drug, physical mixture, formulations and empty microspheres [30]
2. Particle size distribution of prepared microspheres:[31]
The size of the prepared microspheres can be measured by the optical microscopy method using a calibrated stage micrometer for randomly selected samples of all the formulations.
Optical microscopy:-This method is used to determine particle size ofmicrospheres by using optical microscope (MeizerOPTIK) The measurement is done under 45x (10x eyepiece and 45x objective) and100 particles are calculated.
3. Surface topography by Scanning Electron Microscopy (SEM):- SEM of the microspheres shows the surface morphology of the microspheres like their shape and size.
Scanning electron microscopy (SEM):-Surface morphology of microspheres is determined by the method SEM. In this method microspheres are mounted directly on the SEM sample slub with the help of double sided sticking tape and coated with gold film under reduced pressure. Scanning Electron photomicrographs of drug?loaded microspheres are taken. A small amount of microspheres s spread on gold stub. Afterwards, the stub containing the sample is placed in the Scanning electron microscopy (SEM). A Scanning electron photomicrograph is taken at an acceleration voltage of 20KV and chamber pressure of 0.6 mm Hg [31].
Particle size analysis:- The particle sizes and particles size distributions are further analyzed by using dynamic light scattering technique, Microspheres are dispersed into 100 ml of water and sonicated for 1 min to remove agglomerations. The mean volume diameter (Vd) is recorded and polydispersity is determined by the SPAN factor. A high value of SPAN indicates a wide distribution in size and a high polydispersity.
4. Swelling Index:[32]
Swelling index is determined by measuring the extent of swelling of microspheres in a particular solvent. To ensure the complete equilibrium, exactly weighed 100 mg of microspheres ares allowed to swell in solvent for 34 hrs. The excess surface adhered liquid drops are removed by blotting and the swollen microspheres are weighed by using microbalance. The Hydrogel microspheres then dried in an oven at 60° for 5 hrs. until there is no change in the dried mass of sample. The swelling index of the microsphere is calculated by using the formula:
Swelling index =mass of swollen microspheres – mass of dried microsphere/mass of dried × 100
5. Entrapment efficiency:[33]
Microspheres containing of drug (5mg) were crushed and then dissolved in distilled water with the help of ultrasonic stirrer for 3 hr., and was filtered then assayed by uv-vis spectroscopy. Entrapment efficiency is equal to ratio of actual drug content to theoretical drug content.
% Entrapment = Actual content/Theoreticalcontent x 100Swelling.[33]
6. Stability studies:[34]
By placing the microspheres in screw capped glass container and stored them at following conditions:
1. Ambient humid condition
2. Room temperature (27+/-2 0C)
3. Oven temperature (40+/-2 0C)
4. Refrigerator (5 0C -80C).
It was carried out of a 60 days and the drug content of the microsphere was analysed.[34]
7. Density determination:[3]
The density of the microspheres can be measured by using a multi volume pychnometer. Accurately weighed sample in a cup is placed into the multi volume pychnometer. Helium is introduced at a constant pressure in the chamber and allowed to expand. This expansion results in a decrease in pressure within the chamber. Two consecutive readings of reduction in pressure at different initial pressure are noted. From two pressure readings the volume and hence the density of the microsphere carrier is determined.[3]
8. Isoelectric point:[3]
The micro electrophoresis is an apparatus used to measure the electrophoretic mobility of microspheres from which the isoelectric point can be determined. The mean velocity at different PH values ranging from 3-10 is calculated by measuring the time of particle movement over a distance of 1 mm. By using this data the electrical mobility of the particle can be determined. The electrophoretic mobility can be related to surface contained charge, ionisable behaviour or ion absorption nature of the microspheres.[3]
9. Bulk density:[35]
The microspheres fabricated are weighed and transferred to a 10-ml glass graduated cylinder. The cylinder is tapped using an autotrap until the microsphere bed volume is stabilized. The bulk density is estimated by the ratio of microsphere weight to the final volume of the tapped microsphere bed.[35]
10. Angle of contact:[36]
The angle of contact is measured to determine the wetting property of a micro particulate carrier. It determines the nature of microspheres in terms of hydrophilicity or hydrophobicity. The angle of contact is measured at the solid/air/water interface. The angles of contact are measured by placing a droplet in a circular cell mounted above objective of inverted microscope. Contact angle is measured at 200C within a minute of deposition of microspheres.[36]
11. X-ray diffraction:[1]
Change in crystalinity of drug can be determined by this technique. Microperticles and its individual components were analysed by the help of D & discover (Bruker, Germony).Scanning range angle between 8 0C - 70 0C. Scan speed - 4o/min Scintillation detector Primary silt=1mm, Secondary silt=0.6 mm. Thermal analysis of microcapsule and its component can be done by using-
o Differential scanning calorimetry (DSC)
o Thermo gravimetric analysis (TGA)
o Differential thermometric analysis (DTA)
Accurately the sample was weighed and heated on alumina pan at constant rate of 10oc/min under nitrogen flow of 40 ml/min.[1]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Drug release: [37]
In vitro methods
In vitro drug release studies have been employed as a quality control procedure in pharmaceutical production, in product development etc. Sensitive and reproducible release data derived from physic-chemically and hydro dynamically defined conditions are necessary, however no standard in vitro method has yet been developed. Different workers have used apparatus of varying designs and under varying conditions, depending on the shape and application of the dosage form developed.
Beaker method
The dosage form in this method is made to adhere at the bottom of the beaker containing the medium and stirred uniformly using over head stirrer. Volume of the medium used in the literature for the studies varies from 50-500 ml and the stirrer speed form 60-300 rpm.
Interface diffusion system
This method is developed by Dearden & Tomlinson. It consists of four compartments. A represents the oral cavity, and initially contained an appropriate concentration of drug in a buffer. The compartment B representing the buccal membrane, contained 1-octanol, and compartment C representing body fluids, contained 0.2 M HCL. The compartment D representing protein binding also contained 1-octanol. Before use, the aqueous phase and 1-octanol were saturated with each other. Samples were withdrawn and returned to compartment A with a syringe.
Modified Keshary Chien Cell
A specialized apparatus was designed in the laboratory. It comprised of a Keshary Chien cell containing distilled water (50ml) at 370 C as dissolution medium. TMDDS (Trans Membrane Drug Delivery System) was placed in a glass tube fitted with a 10# sieve at the bottom which reciprocated in the medium at 30 strokes per min.
Dissolution apparatus
Standard USP or BP dissolution apparatus have been used to study in vitro release profiles using rotating elements, paddle and basket. Dissolution medium used for the study varied from 100- 500 ml and speed of rotation from 50-100 rpm.
In vivo methods
Methods for studying the permeability of intact mucosa comprise of techniques that exploit the biological response of the organism locally or systemically and those that involve direct local measurement of uptake or accumulation of penetrate at the surface. The most widely used methods include in vivo studies using animal models, buccal absorption tests, and perfusion chambers for studying drug permeability.
Animal models
Animal models are used mainly for the screening of the series of compounds, investigating the mechanisms and usefulness of permeation enhancers or evaluating a set of formulations.Animal models such as the dog, rats, rabbits, cat, hamster, pigs, and sheep have been reported. In general, the procedure involves anesthetizing the animal followed by administration of the dosage form. In case of rats, the oesophagus is ligated to prevent absorption pathways other than oral mucosa. At different time intervals, the blood is withdrawn analyzed.
Buccal absorption test
The buccal absorption test was developed by Beckett & Triggs in 1967. It is a simple and reliable method for measuring the extent of drug loss of the human oral cavity for single and multi component mixtures of drugs. The test has been successfully used to investigate the relative importance of drug structure, contact time, initial drug concentration and Ph of the solution while the drug is held in the oral cavity.
In vitro-In vivo correlations
Correlations between in vitro dissolution rates and the rate and extent of availability as determined by blood concentration and or urinary excretion of drug or metabolites are referred to as “in vitro-in vivo correlations”. Such correlations allow one to develop product specifications with bioavailability.
Percent of Drug Dissolved In Vitro Vs Peak Plasma Concentration
One of the ways of checking the in vitro and in vivo correlation is to measure the percent of the drug released from different dosage forms and also to estimate the peak plasma concentrations achieved by them and then to check the correlation between them.
Percent of Drug Dissolved Vs Percent of Drug Absorbed
If the dissolution rate is the limiting step in the absorption of the drug, and is absorbed completely after dissolution, a linear correlation may be obtained by comparing the percent of the drug absorbed to the percent of the drug dissolved. If the rate limiting step in the bioavailability of the drug is the rate of absorption of the drug, a change in the dissolution rate may not be reflected in a change in the rate and the extent of drug absorption from the dosage form.
Dissolution Rate Vs Absorption Rate
The absorption rate is usually more difficult to determine than the absorption time. Since the absorption rate and absorption time of a drug are inversely correlated, the absorption time may be used in correlating the dissolution data to the absorption data. In the analysis of in vitro and in vivo drug correlation, rapid drug absorption may be distinguished from the slower drug absorption by observation of the absorption time for the dosage form. The quicker the absorption of the drug the less is the absorption time required for the absorption of the certain amount of the drug. The time required for the absorption of the same amount of drug from the dosage form is correlated.
Percent of Drug Dissolved Vs Serum Drug Concentration
For drugs whose absorption from GIT is dissolution rate limited, a linear correlation may be established between the percent of drug dissolved at specified times and the serum drug concentrations at corresponding times.
Percent of Drug Dissolved Vs Percent of the Dose Excreted in urine
The percent of a drug dissolved and the percent of drug absorbed are linearly correlated. There exists a correlation between the amount of drug in body and the amount of drug excreted in the urine. Therefore, a linear relation may be established between the percent of the drug dissolved and the percent of the dose excreted in the urine.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
APPLICATIONS: [38],[39],[40],[41]
New applications for microspheres are discovered every day, below are just a few:
Assay - Coated microspheres provide measuring tool in biology and drug research.
Buoyancy - Hollow microspheres are used to decrease material density in plastics (Glass and polymer).
Ceramics - Used to create porous ceramics used for filters (microspheres melt out during firing, Polyethylene Microspheres).
Cosmetics - Opaque microspheres used to hide wrinkles and give color, Clear microspheres provide "smooth ball bearing" texture during application (Polyethylene Microspheres).
Drug delivery - As miniature time release drug capsule made of, for example, polymers. A similar use is as outer shells of micro bubble contrast agents used in contrast-enhanced ultrasound.
Electronic paper - Dual Functional microspheres used in Gyricon electronic paper.
Personal Care - Added to Scrubs as an exfoliating age(Polyethylene Microspheres).
Spacers - Used in LCD screens to provide a precision spacing between glass panels (glass).
Standards - monodispere microspheres are used to calibrate particle sieves, and particle counting apparatus.
Retroreflective - added on top of paint used on roads and signs to increase night visibility of road stripes and signs (glass).
Thickening Agent - Added to paints and epoxies to modify viscosity and buoyancy.
Cancer research: [38]
One useful discovery made from the research of microspheres is a way to fight cancer on amolecular level. According to Wake Oncologists, "SIR-Spheres microspheres are radioactivepolymer spheres that emit beta radiation. Physicians insert a catheter through the groin intothe hepatic artery and deliver millions of microspheres directly to the tumor site. The SIR Spheresmicrospheres target the liver tumors and spare healthy liver tissue. Cancermicrosphere technology is the latest trend in cancer therapy. It helps the pharmacist toformulate the product with maximum therapeutic value and minimum or negligible range sideeffects. A major disadvantage of anticancer drugs is their lack of selectivity for tumor tissue alone, which causes severe side effects and results in low cure rates. Thus, it is very difficult to target abnormal cells by the conventional method of the drug delivery system. Microsphere technology is probably the only method that can be used for site-specific action, without causing significant side effects on normal cells.
Recent Applications of Controlled Release Microspheres
Controlled-Release Vaccine
Vaccination has been highly successful for controlling or even eradicating many importanttypes of infectious diseases, and new or improved vaccines are being heavily investigated forAIDS,hepatitis B, anthrax, and SARS. A frequent problem is the need for repeatedadministrations—usually KYEKYOON “KEVIN” KIM AND DANIEL W. PACKinjections—to ensure long-lasting immunity. For example, the current anthrax vaccinerequires a series of boosters at 2 and 4 weeks, and at 6, 12, and 18 months following the firstinoculation; and the Recombivax H-Brvaccine for hepatitis B—required for most healthcareworkers in the U.S.—is administered in three injections at 0, 1, and 6 months. The need formultiple injections poses a serious problem for patients in developing countries with limited access to medical care, where awareness is lacking, or for transient populations.






