
About Author:
Karthika M
Postgraduate in Bioinformatics
Bharathiar University
m.karthika1@gmail.com
ABSTRACT
Hepatitis C virus (HCV) is a major human pathogen associated with life-threatening liver disease, affect 170 million people worldwide. Hepatitis C virus (HCV) envelope proteins mediate the entry of virus into cells by binding to cellular receptors, resulting in fusion of the viral membrane with the host cell membrane and permitting the viral genome to enter the cytoplasm. Entry inhibitors would inhibit the viral entry itself.
Envelope protein E2, responsible for viral entry taken as target is modeled using Modeller 9.10. 47 small molecules have been taken for docking with envelope protein.10 small molecules have been selected for Induced Fit Docking based on the Glide energy, Docking Score.
Ligand 20 is showing good binding affinity towards the target in active site, having Docking Score -6.07, Glide Energy -60.41, IC50 value 1.5 and it obeys Lipinski’s Rule of Five. ADMET Properties were also studied for these compounds. All computational works are performed in the Schrodinger suite 2009. Hence the present study suggests that the ligand 20 with further in vitro and in vivo testing can be introduced as an effective inhibitor against HCV for development of curative therapy against Hepatitis C.
REFERENCE ID: PHARMATUTOR-ART-1650
INTRODUCTION
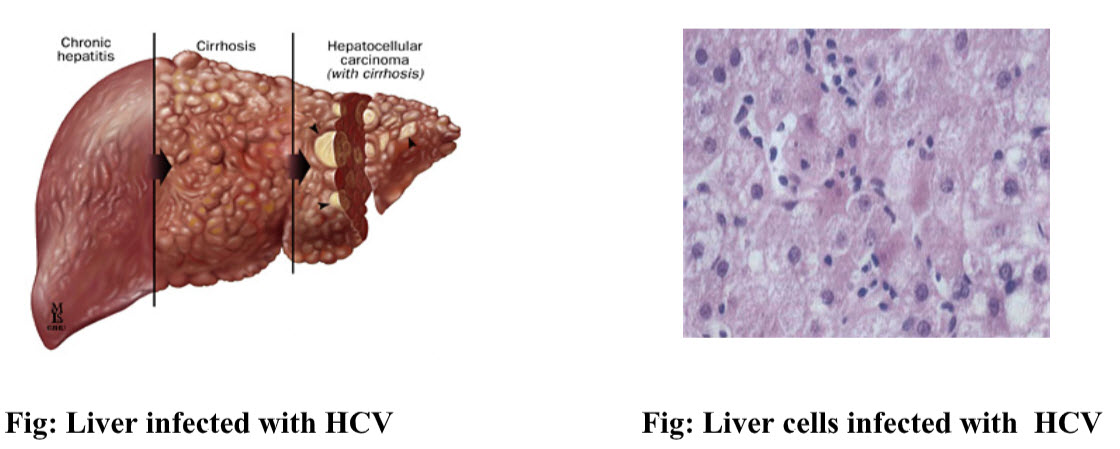
Hepatitis C is a contagious liver disease that results from infection with hepatitis C virus (HCV). The infection is often asymptomatic, but chronic infection can lead to scarring of the liver and ultimately to liver cirrhosis. In some cases, those with cirrhosis will go on to develop liver failure, liver cancer or life-threatening esophageal and gastric varices. HCV is transmitted through contact with the blood of an infected person. (Jou, Muir, 2008)
The Hepatitis C virus persists in the liver in about 85% of those infected. This persistent infection can be treated with medication; peginterferon and ribavirin are the current standard therapy. Overall, between 50–80% of people treated are cured. Those who develop cirrhosis or liver cancer may require a liver transplant. Hepatitis C is the leading cause of liver transplantation though the virus usually recurs after transplantation. No vaccine against hepatitis C is currently available.(Ghany MG et al.,2009)
Hepatitis C virus(HCV)
Hepatitis C virus (HCV) is a small (55–65 nm in size), enveloped, positive-sense single-stranded RNA virus of the family Flaviviridae. Hepatitis C virus is the cause of hepatitis C in humans.(Kapoor et al.,2011)
Structure
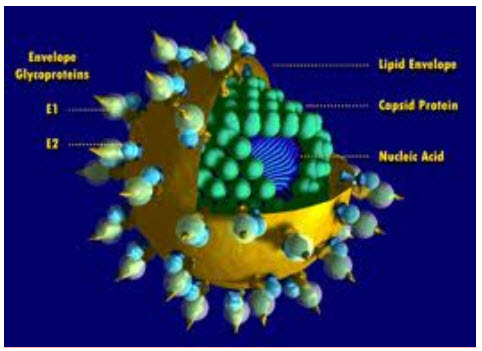
Fig : Structure of Hepatitis C Virus
The hepatitis C virus particle consists of a core of genetic material (RNA), surrounded by an icosahedral protective shell of protein, and further encased in a lipid (fatty) envelope of cellular origin. Two viral envelope glycoproteins, E1 and E2, are embedded in the lipid envelope.
Genome
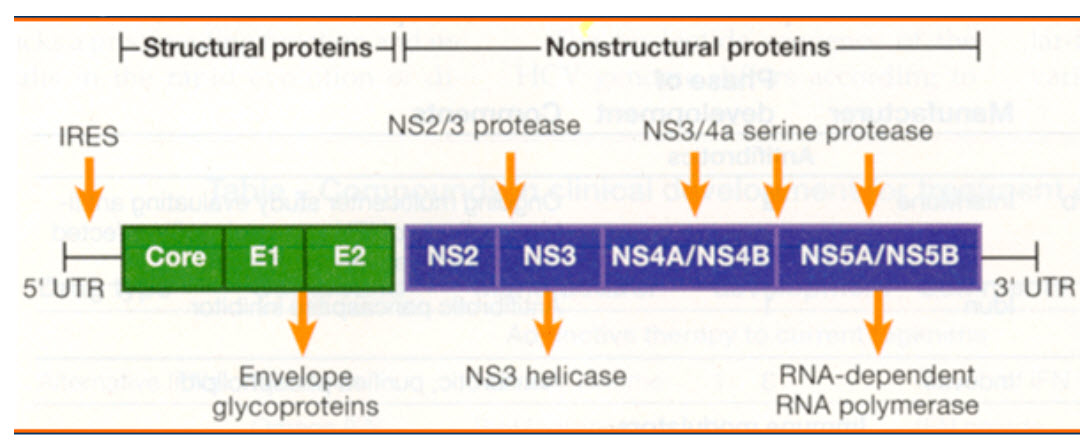
Fig: HCV Genome
HCV Envelope Protein as a Target
Cellular processes, such as clathrin-mediated endocytosis or endosome formation, are probably not very clean targets, and agents designed to target those processes would be likely to have severe adverse effects. Agents that bind host factors may interfere with normal cellular functions, resulting in an unacceptable adverse effect profile. Agents that bind viral components in the entry process, such as the HCV envelope proteins, might be more likely to be selected as target.
Entry Inhibitors
Entry inhibitors may be particularly helpful after liver transplantation, where universal reinfection of the graft often results in rapid fibrosis progression and subsequent graft failure. Even therapies transiently inhibiting HCV cell entry could prevent graft reinfection and thus greatly improve the effectiveness of liver transplantation.
MATERIALS AND METHODS
Molecular Modelling
This procedure, also termed Comparative Modelling or knowledge based modelling developed a three-dimensional model from a protein sequence (target) based on the structures of homologous proteins (Templates).
Modeller
Modeller is a computer program that model three-dimensional structures of proteins and their assemblies by satisfaction of spatial restraints.
The user provided an alignment of a sequence to be modeled with known related structures and Modeller automatically calculated a model containing all non-hydrogen atoms.More generally, the input to the program was restraints on the spatial structure of the amino acid sequence and ligands to be modeled. The output was a 3D structure that satisfied these restraints as well as possible. A 3D model was obtained by optimization of a molecular Probability Density Function (PDF). The molecular PDF for comparative modeling was optimized with the variable target function procedure in Cartesian space that employed methods of conjugate gradients and molecular dynamics with simulated annealing.
Searching for Structures
First, it was necessary to put the target sequence into the PIR format readable by Modeller.
A search for potentially related sequences of known structure could be performed by the profile.build() command of MODELLER.
Selecting a Template
To select the most appropriate template for the query sequence over the similar structures, we would use thealignment.compare_structures() command to assess the structural and sequential similarity between the possible templates.
Model Building
Once a target-template alignment was constructed, MODELLER calculated a 3D model of the target completely automatically, using its automodel class. The following script would generate five similar models of HCV E2 based on the template structure and the alignment file “align_HCV.ali”. The most important output files were ‘model-single-log’, which reported warnings, errors and other useful information including the input restraints used for modeling that remain violated in the final model, which contained the coordinates of the five produced models, in the PDB format.
Model Evaluation
If several models were calculated for the same target, the ‘best’ model could be selected by picking the model with the lowest value of the MODELLER objective function. The DOPE potential in MODELLER was used to evaluate the model fold.
Molecular Docking
The computational process of searching for a ligand that is able to fit both geometrically and energetically to the binding site of a protein is called molecular docking.
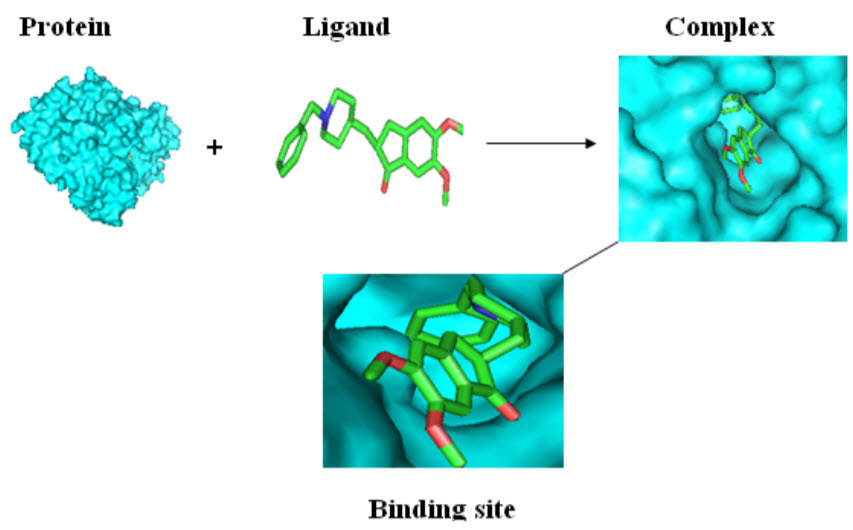
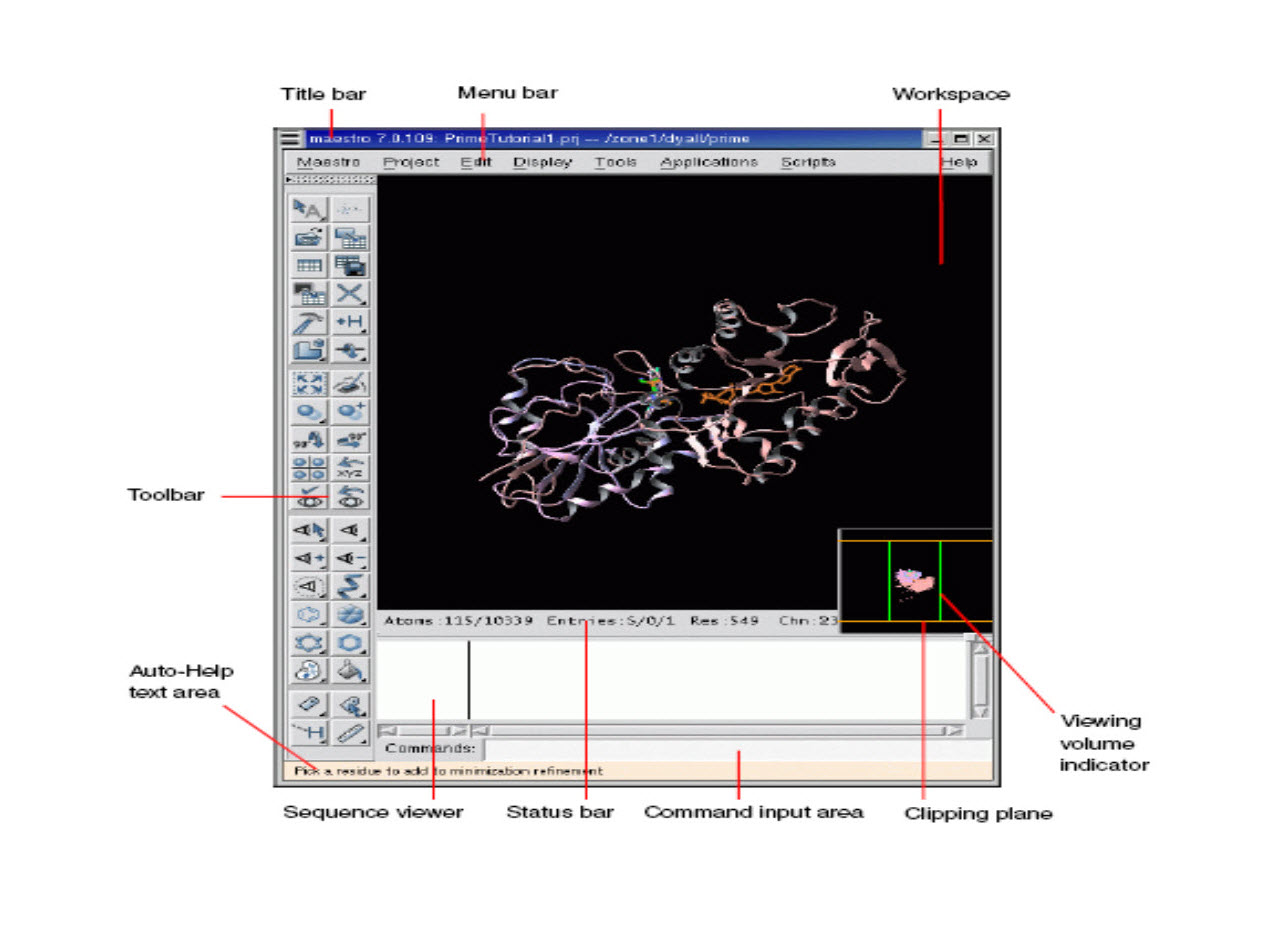
Docking Analysis using MAESTRO
Maestro is the graphical user interface for all of Schrodinger’s products like CombiGlideTM, EpikTM, GlideTM, ImpactTM, LiaisonTM, LigprepTM,MacroModelTM,PhaseTM, PrimeTM, QikPropTM, QsiteTM, and StrikeTM. It contains tools for building, displaying, and manipulating chemical structures for organizing, loading and storing these structures and associated data, and for setting up, monitoring, and visualizing the results of calculations on these structures.
Docking Method – GLIDE(Grid Based Ligand Docking with Energitics)
Glide searches for favorable interactions between one or more typically small ligand molecules and a typically larger receptor molecule usually a protein. Each ligand must be a single molecule, while the receptor may include more than one molecule such as a protein and a cofactor. GLIDE can be run in rigid or flexible docking modes and it the later automatically generates conformation for each input ligand. The combination of positions and orientation of the ligand relative to the receptor along with its conformation in flexible docking is referred to as a ligand pose.
Protein Preparation
A typical PDB structure file consists only of heavy atoms. Therefore, hydrogen does have to be added prior to use in Glide calculations, which use an all-atom force field.
The protein preparation facility consists of two components, preparation and refinement. After ensuring chemical correctness, the preparation component adds hydrogen and neutralizes side chains that are not close to the binding cavity and do not participate in salt bridges. The refinement component performs a restrained impact minimization of the co-crystallized complex, which reorients side-chain hydroxyl groups and alleviates potential steric clashes.
Ligand Preparation
Glide also allows importing the lead molecule in SDF, mol or mol2 format which is drawn using chemsketch software. These determined structures are minimized using two methods
1. Ligprep Minimization
2. Impact Minimization
Ligprep generate tautomers and conformers for single ligand and Impact minimization uses two algorithms for minimization steepest descent and conjugate gradient which runs for 500 to 1000 cycles. Both methods use OPLS force field for minimizing the structures.
Receptor Grid Generation
The ligand should be selected for the ligand to identify the active site to bind with the protein.
If the protein structure in PDB is not complexed with ligand, then the active site of the protein should be specified. We can particularly specify the residues or can specify the region.
Ligand Docking
For docking the ligand with the target protein the receptor grid file should be specified and the ligprep out file should be selected.
Designing of Inhibitors
Novel class of HCV Rimcazole derivatives were recently reported for their anti-HCV activity and they were designed using MARVINSKETCH and PYMOL. Totally 47 Rimcazole derivatives were modeled using MarvinSketch software.
Collection of protein
Protein is retrieved from PDB (protein data bank) based on the lowest resolution. To model the protein, the template structure is selected (PDB id : 2xfc) from PDB.
Modelled Structure of HCV E2
Protein Information
|
Length |
862 Residues |
|
Target Sequence |
Uniprot ID – Q99IB8 |
|
Template |
Chikungunya Virus Envelope Protein E2 PDB ID – 2XFC |
|
Active Site Residues |
Lys 373, Asn 409, Trp 414, Asp 417, Glu 418, Leu 705, Tyr 706, Asp707, Ser 708, Ser 710, Thr 712, Val 714. |
RESULTS
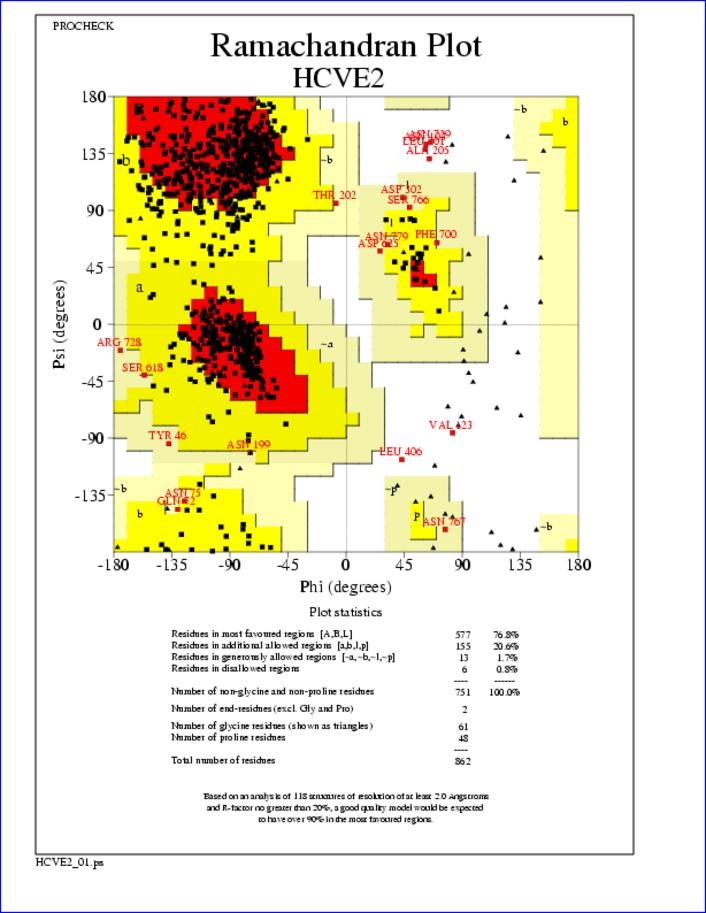
Ramachandran Plot for HCV E2 Glycoprotein
Docking Result
|
Compounds |
Docking Score |
Glide Energy(kcal/mole) |
|
20 |
-4.01 |
-51.50 |
|
19 |
-5.32 |
-49.91 |
|
34 |
-5.35 |
-49.75 |
|
6 |
-5.51 |
-49.72 |
|
7 |
-4.8 |
-49.65 |
|
4 |
-6.24 |
-48.84 |
|
3 |
-6.24 |
-48.84 |
|
5 |
-6.24 |
-48.84 |
|
30 |
-4.49 |
-48.28 |
|
27 |
-3.92 |
-47.32 |
|
8 |
-5.08 |
-46.68 |
|
22 |
-4.74 |
-46.56 |
|
12 |
-4.01 |
-46.55 |
|
28 |
-4.40 |
-46.28 |
|
33 |
-5.28 |
-46.20 |
|
35 |
-4.82 |
-45.55 |
|
18 |
-4.58 |
-45.39 |
|
14 |
-4.69 |
-45.21 |
|
41 |
-4.83 |
-44.95 |
|
9 |
-4.88 |
-44.63 |
|
26 |
-3.62 |
-44.62 |
|
46 |
-4.55 |
-44.51 |
|
44 |
-4.55 |
-44.51 |
|
45 |
-4.55 |
-44.51 |
|
43 |
-4.55 |
-44.51 |
|
47 |
-4.55 |
-44.51 |
|
42 |
-4.55 |
-44.51 |
|
31 |
-5.32 |
-44.21 |
|
23 |
-5.26 |
-43.89 |
|
32 |
-4.44 |
-42.59 |
|
10 |
-4.12 |
-41.70 |
|
40 |
-6.58 |
-41.40 |
Induced Fit Docking Results
|
Ligands |
Docking Score |
Glide Energy (kcal/mole) |
Interaction |
Distance (Å) |
|
20 |
-6.07 |
-60.41 |
[N-H…O] Tyr367 Lys373[N-H…O] |
3.043 2.986 |
|
25 |
-6.84 |
-56.73 |
Lys373[N-H…O] Lys574[N-H…O] |
3.017 2.803 |
|
30 |
-8.19 |
-55.52 |
Asn409 [N-H…O] Gln418 [N-H…O] |
3.022 2.848 |
|
24 |
-6.08 |
-54.17 |
Lys373[N-H…O] Lys574[N-H…O] |
2.918 3.089 |
|
34 |
-7.85 |
-54.01 |
[N-H…O] Asp417 Asp417 [N-H…O] Asn409 [N-H…O] Trp414 [N-H…O] |
3.796 3.130 2.867 2.852 |
Induced Fit Docking Results of Ligand 20
|
Poses |
Docking Score |
Glide Energy (kcal/mole) |
Interaction |
Distance (Å) |
|
1 |
-6.07 |
-60.41 |
[N-H…O] Tyr367 Lys373[N-H…O] |
3.043 2.986 |
|
2 |
-5.70 |
-55.90 |
Lys373 [N-H…O] Lys371 [O-H…O] |
2.980 3.044 |
|
3 |
-4.26 |
-54.70 |
Lys373 [N-H…O] Lys574 [N-H…O] [N-H…O] Glu577 |
2.992 2.802 |
|
4 |
-8.19 |
-55.52 |
Asn409 [N-H…O] Gln418 [N-H…O] |
3.022 2.848 |
|
5 |
-6.30 |
-54.42 |
[N-H…O] Glu577 Lys574 [N-H…O] Lys373 [N-H…O] |
2.909 3.065 2.732 |
|
6 |
-7.44 |
-53.27 |
Gln418 [N-H…O] |
2.895 |
|
7 |
-7.62 |
-53.13 |
Gln418 [N-H…O] Asn409 [N-H…O] |
2.904 3.214 |
Interaction of Ligand 20 in PYMOL
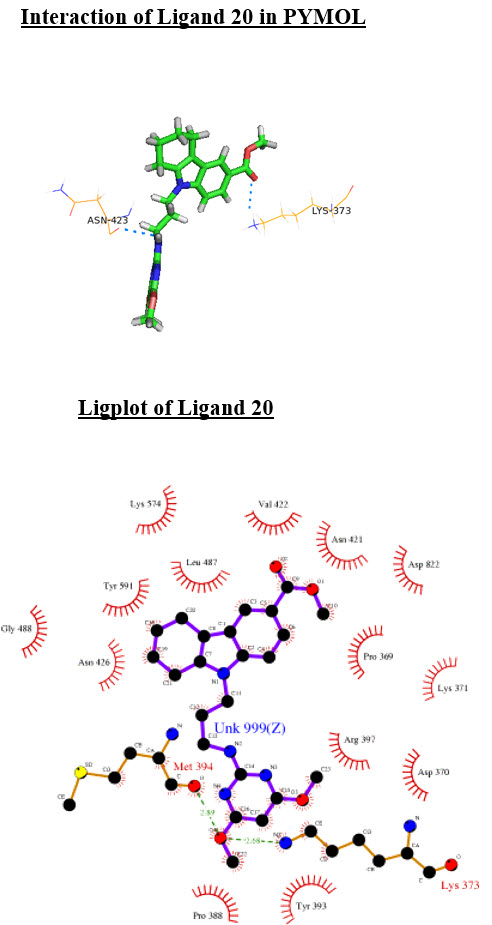
Ligplot of Ligand 20
DISCUSSION
The compound 20 is most potent with an IC50 of 1.5 nM because of the presence of methyl ester group in the series of compounds. The methyl ester at the 3-position improved the potency by several fold. The dimethyl sulfonamide compound 15 showed excellent potency with an IC50 of 5 nM. The N-methylated analogue 24 is as potent as compound 20. When compared to the corresponding 4,6-disubstituted-2-amino pyrimidines, aromatic heterocycles/aromatics such as phenyl sulfone (30) or difluoro phenyl sulfonamides (34) are also active but less potent than pyrimidine analogues. Disubstituted 2-amino/2-oxo pyrimidines and dimethyl sulfonamide groups are best for high potency. Based on binding competition and resistance profiles, this compound class appears to be mechanistically similar to ITX 5061.
CONCLUSION
Envelope Protein E2 (Glycoprotein) responsible for HCV Entry is targeted to inhibit the HCV. Both protein and ligands are minimized using OPLS forcefield. From the 47 compounds, 10 compounds were selected for Induced Fit Docking based on Docking Score and Glide Energy. The ligands chosen abides Lipinski’s Rule of Five. These small molecules belong to a distinct chemical class from ITX 5061, first in class drug, currently in Phase Ib Clinical Trial.
Ligand 20 is showing good binding affinity towards the target having Docking Score -6.07 and Glide Energy -60.41 and IC50 value 1.5nM and it binds with the Active site of the target protein. Hence the present study suggests that the ligand 20 with further in vitro and in vivo testing can be introduced as an effective inhibitor against HCV for development of curative therapy against Hepatitis C.
References
1) Kato N (2000). "Genome of human hepatitis C virus (HCV): gene organization, sequence diversity, and variation". Microb. Comp. Genomics 5 (3): 129–51.
2) Op De Beeck A, Dubuisson J (2003). "Topology of hepatitis C virus envelope glycoproteins". Rev. Med. Virol. 13 (4): 233–41.
3) F. Habersetzer, A. Fournillier, J. Dubuisson, D. Rosa, S. Abrignani, C. Wychowski, I. Nakano, C. Trépo, C. Desgranges, G. Inchauspé, Characterization of human monoclonal antibodies speci?c to the hepatitis C virus glycoprotein E2 with in vitro binding neutralization properties, Virology 249 (1998) 32–41.
4) Targeting HCV entry for development of therapeutics. Wong-Staal F, Syder AJ, McKelvy JF.Viruses. 2010 Aug;2(8):1718-33. Epub 2010 Aug 18.
5) Glycosylation of hepatitis C virus envelope proteins. Goffard A, Dubuisson J. CNRS-UPR2511, Institut de Biologie de Lille, Institut Pasteur deLille, Lille, France. Biochimie. 2003Mar-Apr;85(3-4):295-301.
6) Z.Y. Keck, A. Op de Beeck, K.G. Hadlock, J.M. Xia, T.K. Li, J. Dubuisson, S.K.H. Foung, Hepatitis C virus E2 has three immunogenic domains containing conformational epitopes with distinct properties and biological functions, J. Virol. 78 (2004)9224–9232.
7) G. Leroux-Roels, R. DeLeys, L. Stuyver, A.Elewaut, J. Philippé, I. Desombere, J.Paradijs, G. Maertens, Lymphoproliferative responses to hepatitis C virus core, E1, E2, and NS3 in patients with chronic hepatitis C infection treated with interferon alfa, Hepatology 23 (1996) 8–16.
8) B. Bartosch, J. Dubuisson, F.L. Cosset, Infectious hepatitis C virus pseudo- particles containing functional E1–E2 envelope protein complexes, J. Exper.Med. 197 (2003) 633–642.
9) M. Rodriguez-Rodriguez, D. Tello, B. Yelamos, J. Gomez-Gutierrez, B. Pacheco, S. Ortega, A.G. Serrano, D.L. Peterson, F. Gavilanes, Structural properties of the ectodomain of hepatitis C virus E2 envelope protein, Virus Res. 139 (2009) 91–97.
10) V. Sandrin, P. Boulanger, F. Penin, C. Granier, F.L. Cosset, B. Bartosch, Glycosylation of hepatitis C virus envelope proteins, Biochimie 85 (2003) 295–301.
11) A.OpDe Beeck, L. Cocquerel, J. Dubuisson, Biogenesis of hepatitis C virus glycoproteins, J. Gen. Virol. 86 (2005) 3189–3199.
12) A. Op De Beeck, C. Voisset, B. Bartosch, Y. Ciczora, L. Cocquerel, Z. Keck, S. F. Helle, J. Dubuisson, Hepatitis C virus entry into host cells,Cell Mol. Life Sci. Cell entry of hepatitis C virus, Virology 348 (2006) 1–9.
13) M. Lambot, S. Fretier, A.O. De Beeck, B. Quatannens, S. Lestavel, W. Clavey, J. Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633-642.
14) Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633-642.
15) Bankwitz D., Steinmann E., Bitzegeio J., Ciesek S., Friesland M., Herrmann E., Zeisel M. B., Baumert T. F., Keck Z. Y., et al., Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol 84, 5751–5763.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








