
About Authors: Aswini K. Reddy, Swetha Yasa, Srivally Challa, Masoom. md
Mallareddy Institute of Pharmaceutical Sciences, Hyderabad
ABSTRACT
Both solid- and liquid-phase combinatorial chemistry have emerged as powerful tools for identifying pharmacologically active compounds and optimizing the biological activity of a lead compound. Complementary high-throughput in vitro assays are essential for compound evaluation. Cell-based assays that use optical endpoints permit investigation of a wide variety of functional properties of these compounds including specific intracellular biochemical pathways, protein-protein interactions, and the subcellular localization of targets. Integration of combinatorial chemistry with contemporary pharmacology now represents an important factor in drug discovery and development.
This is an exceptionally exciting time in the field of pharmacology. The environment for the identification of new therapeutic targets and agents that interact with these targets has rapidly changed with the application of genetic tools and genomics. Extrapolation from the genomic sequencing of lower organisms suggests that there will be a 10-fold increase in the number of potential human therapeutic targets in the next several years with the completion of the Human Genome Project (Drews, 1996). This is leading to a fundamental transformation in pharmacology; no longer is there a dearth of molecular targets for small molecules. Rather, the emphasis is now on validating whether or not the targets are appropriate for therapeutic intervention, on generating large arrays of compounds that represent diverse portions of “chemical space”, and developing methods to quickly assess the credentials of small molecules as target disrupters. We believe many of the tools and reagents that are being developed to facilitate this scientific activity will emerge as vital for future academic pharmacological research. Perhaps most important will be the exploitation of combinatorial chemistry libraries, which are becoming widely available. Although we cannot comprehensively review this broad topic here, the goal of this brief commentary is to portray some of the strategies and potentials of combinatorial chemistry libraries as they relate to pharmacological studies.
[adsense:336x280:8701650588]
Reference ID: PHARMATUTOR-ART-1088
Discussion
Combinatorial Chemistry Strategies. Combinatorial chemistry has emerged because of genomics and new efficient biological screening strategies and is widely cited as a paradigm shift in the way that new small molecule lead structures will be identified (Choong and Ellman, 1996; Gold and Alper, 1997; Obrecht and Villalgordo, 1998). Traditionally, natural product extracts or industrial collections of randomly synthesized organic molecules were screened to discover “hits”, which were then optimized in an iterative process by the synthesis of derivatives. Combinatorial synthesis (Curran and Wipf, 1997) is now supplementing traditional strategies at both the lead discovery and the lead optimization stage. Surveys of very recent combinatorial chemistry-derived structure activity relationships serve to underline the tremendous impact that this technology already has had on drug discovery in industry and academe (Dolle and Nelson, 1999). Combinatorial synthesis assembles building blocks to make new molecules. Large libraries (.100,000) can be made by assembling all possible combinations of a set of building blocks, and new lead compounds can be identified by using techniques like split-mix synthesis (Gallop et al., 1994; Gordon et al., 1994; Nefzi et al., 1997) coupled with suitable tagging or deconvolution strategies (Nestler et al., 1994) . Smaller discovery libraries (,10,000) can be made as individual components by rapidly advancing techniques of Received for publication December 6, 1999. automated parallel synthesis (Hogan, 1996), and the further optimization of a lead discovered in a combinatorial process follows naturally because the building blocks are already in place . Focused (directed screening) or secondary libraries have a more narrowly defined structural focus and carve out structure-activity relationships for increasing potency and selectivity that are critical in a lead molecule. In this strategy, the synthesis of tailored building blocks is often the rate-limiting step, and the synthetic sequence can exceed eight steps and tolerate some low-yielding methodology; parallel purification techniques and other specialized combinatorial chemistry methods can be used. Most of the commercial automation vendors are now aggressively targeting these applications where sample size remains in the 100s or low 1000s. Ideally, products will be well characterized and of high purity, but very often an arbitrary purity cutoff of 80% or higher will suffice to limit the number of “false positive” hits. Lower purities, which often are the result of incomplete reactions, can be tolerated if the structure of the major side products is known.
The lead(s) may come from an existing program, from a screening hit from a discovery library or a compound database (including virtual libraries), from the literature or from a competitor’s compound, or from a rational design structurebased approach. The integration of structure-guided design and combinatorial chemistry is referred to as the targeted diversity approach. The ArQule group has used this approach for the successful preparation of a library of HIV protease inhibitors (Hogan, 1997). Structurally complex natural products form a particularly attractive resource for targeted array design of exploratory libraries. For example, a library of dual-specificity phosphatase inhibitors was based on the structures of the natural product serine-threonine protein phosphatase inhibitors microcystin LR and calyculin A (Fig. 1) (Rice et al., 1997; Wipf et al., 1997). Much of the efficiency of combinatorial synthesis strategies is due to improvements in the work-up of organic reactions by attachment of the substrate molecule to an insoluble resin, most often a polystyrene bead, 100 to 150 mm in diameter,
ABBREVIATIONS: SPOS, solid-phase organic synthesis; FRET, fluorescence resonance energy transfer.
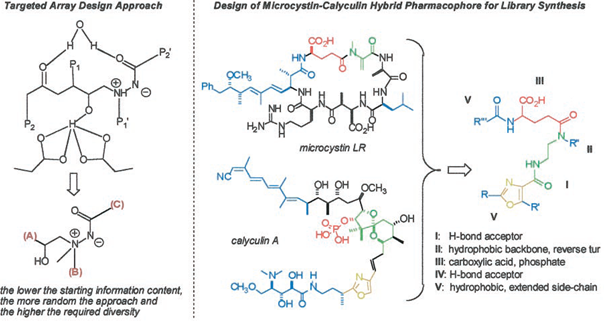
Fig. 1. Library scaffold design. This illustrates how a pharmacophore can be designed from natural products (for additional details, see Rice et al., 1997
and Wipf et al., 1997). copolymerized with divinyl benzene. The technique of solidphase organic synthesis (SPOS) is a logical extension toward small molecule synthesis of the methodology from solidphase peptide (Jung, 1996) and oligonucleotide (Pirrung, 1997) synthesis where supported phase synthesis has emerged as the medium of choice for repetitive couplings of standard building blocks.
[adsense:468x15:2204050025]
A central aim of SPOS is to overcome the unpredictable physical characteristics of synthetic intermediates and products by covalent attachment to a polymeric support. Purification is then facilitated by removal of excess reagents and by-products by filtration. This heterogeneous approach represents the most widely applied variant of the general idea to simplify isolation of pure compounds. Some of the advantages of SPOS include: excess reagents can be used; the reactions can be driven to completion; filtration can be used for rapid purification; automation is easily achieved; and relative site isolation. Among the limitations of SPOS are requirements for linkage or cleavage steps that may restrict the possible chemical reactions, restrained control of reaction stoichiometry, difficulty in purification after low-yielding steps, timeconsuming conversion of solution-phase chemistry to solidphase chemistry, and scale-up restrictions. In part because of the limitations of SPOS, alternative strategies have been developed that exploit the benefits of conducting liquid-phase polymer-supported synthesis in a homogeneous environment. In this approach, separation is accomplished via membrane ultrafiltration, precipitation, or crystallization. More recently, solution-phase methods have emerged to complement solid-phase techniques. To date, most of these methods are founded on acid-base chemistry and sequestration-enabling reagents (Gayo and Suto, 1997; Parlow et al., 1997; Siegel et al., 1997). The pioneering acid-base extraction work of Boger and coworkers (Boger et al., 1996), for example, clearly shows that hundreds to thousands of individual pure molecules can be made by solution synthesis, if the purification methods are simple enough. The use of fluorous tags provides organic compounds with sufficient affinity to perfluorinated solvents that are immiscible with water and standard organic solvents to allow selective purification via fluorous liquid-liquid extraction or fluorinated silica gel (Fig. 3) (Studer et al., 1997). Thus, the advantages of liquid-phase synthesis are that all organic reactions can be used, extensive purification and compound characterization is possible after every step, and large-scale syntheses are possible. Solution-phase parallel synthesis is, however, more labor and time intensive than SPOS, and automation is more difficult. In addition, driving reaction to completion is challenging and repeated chromatographic purification is often necessary. An important aspect of any library synthesis is quality control. Both the identity and purity of the library samples has to be ascertained for all or a representative selection of compounds. Among the currently available methods, liquid chromatography-mass spectroscopy, which has almost become the universal standard, and high-throughput NMR are particularly valuable. IR is an excellent method for on-bead reaction control (Yan, 1998). Gel-phase NMR or magic angle spinning-NMR can also be used for assaying the structure of samples still attached to solid support.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
Principles of Rapid Compound Analyses. Traditional pharmacological approaches using intact animals, ex vivo preparations, and cell-based or cell-free (in vitro) assays readily accommodate analysis of individual compounds. The compound libraries derived from combinatorial chemistry, however, demand high-throughput detection systems to optimize their utility. Mindful that the quantity of each element in the libraries is limiting and not always pure, cellfree, in vitro systems have been the primary screening tools either in a microtiter plate format (96, 384, or 1536 wells per plate) or in a solid or semisolid matrix. An excellent review describing the merits of different automation schemes and assay platforms has been provided elsewhere (Houston and Banks, 1997). It seems likely that the highly popular liquid workstations commonly found in pharmaceutical laboratories will begin to populate academic laboratories as the advantages of rapid screening are appreciated and their price decreases. The most popular format for cell-free highthroughput systems is the 96-well plate because of the availability of a multitude of detection systems. Disruption of enzyme activity or protein-protein interactions can be moni- tored using a variety of endpoints including radioactivity, fluorescence, absorbance, surface plasmon resonance, and immunoreactivity. Of particular concern is the use of only small amounts of reagents, including the test samples, and a relatively rapid but robust assay system. The initial strategy
used to evaluate the compounds usually seeks a qualitative or semiquantitative answer, namely do any of the compounds affect the system in a desirable manner. More refined studies of the nature of the activity are reserved to a time when purer compound and larger quantities are available. Thus, complete concentration-response curves, so highly desired by pharmacologists, often must wait. Reagent composition, such as the assay protein, is often designed for quick isolation and purification. Thus, recombinant fusion proteins with tags that facilitate purification, such as glutathione S-transferase or multiple histidines, are favored. Studies from several laboratories serve to illustrate how such a process would be implemented. Gray et al. (1999), for example, used a 96-well plate solution-phase phosphorylation assay and a combinatorial library synthesis approach with a purine scaffold to generate selective inhibitors of an important cyclin-dependent protein kinase, Cdk2. In another example, potential Cdc25 dual-specificity phosphatase inhibitors from a targeted array library were evaluated using 96-well microtiter plates and recombinant human enzyme fused to glutathione S-transferase (Rice et al., 1997). Targeting Cdc25 dual-specificity phosphatases normally demands the full-length protein substrate, namely a cyclin-dependent kinase, which would need to be made by recombinant techniques and phosphorylated. Thus, a more rapid and inexpensive approach was used with an artificial, commercially available, smallmolecular-weight substrate, which adopted enhanced fluorescent intensity when dephosphorylated. All of the compounds in the library were easily and quickly examined against at least seven protein phosphatases to assess both activity and specificity. Once a potentially active compound was identified, the investigators obtained a more highly purified compound and expanded the evaluation by using complete concentration-response curves and determining the nature and kinetics of the inhibition (Rice et al., 1997). Iterating chemical library synthesis and biological screening should permit the identification of even more potent and efficacious compounds built on this lead structure. More recently, cell-based systems have been developed that greatly expand the pharmacological questions that now can be addressed with combinatorial libraries. Many of the contemporary molecular targets are intracellular and, thus, demand that the test compounds enter cells to act. Cell-based assays permit interrogation of compounds for this property. Moreover, some targets, such as ion channels or transcription factors, inherently require cell-based functional assays to detect disruption. Cell-based assays also permit more information- rich, multiparameter readouts using live cells even in real time. Typically, cell-based assays rely on optical methods for detection (Gonzalez and Negulescu, 1998). Some of the more popular methods are listed in Table 1. A primary advantage of these optical readouts is that the endogenous background signal can be quite small, whereas the signal from the indicator can be very bright. These properties enable the high signal-to-noise and sensitivity required for high-throughput assays. Chemiluminescent assays use chemical reactions to generate light. The most established probes for intracellular detection are the bioluminescent substrate for luciferase, luciferin, which is often used in reporter gene constructs, and the Ca21-sensitive protein aequorin. Substrates for both of these systems can be delivered to intact cells. The primary advantage offered by luminescent indicators is the low background signal and, consequently, luminescence assays are very sensitive and have a large dynamic range. The low photon flux of the current luminescence systems, which is about 0.1 to 0.2 photons per second, however, restricts this method to large cell numbers and can require high intracellular expression that is often only obtained by transient transfection protocols (Gonzalez and Negulescu, 1998; Gonza´ lez et al., 1999). In contrast to chemiluminescence, the photons in fluorescence systems are used to generate an excited state rather than a chemical reaction. The excited state is short-lived and can be repeatedly excited, yielding a bright signal. Thus, single cell assays are possible with spatial resolution sufficient for the investigators to determine the subcellular distribution of the fluorescence probe. Single-cell detection can also be quite useful because it allows separation of expressing cells and, thus, enrichment of the cell population for function transfectants. A potential problem with fluorescence is that the library compounds may have inherent fluorescence that causes false readings.
A particularly powerful fluorescence-based approach is fluorescence resonance energy transfer (FRET), which results when two fluorophores with appropriate spectral overlap are in close proximity either due to intra- or intermolecular interactions. For example, one can even use FRET to detect the conformation and metal binding of proteins, such as metallothionein, within cells (Pearce et al., 2000). Excited energy is transferred from the donor to the acceptor, which results in a decrease in donor intensity and a concomitant increase in acceptor intensity. The ratiometric detection of the changes in signals not only improves reproducibility and sensitivity but it also markedly reduces cell number, probe concentration, and optical path distance as variables. Restrictions of FRET are: the optimal spatial location or orientation of fluorescence probes is often not known; high levels of intracellular probe expression are required and sometime difficult to achieve except with transient transfection; and the spectral overlap between the emissions of the donor and acceptor can reduce the dynamic range of the assay. The power of FRET, especially for mapping the subcellular localization of targets and intracellular protein-protein interactions, makes it likely that its use with combinatorial libraries will increase as more probes are developed.
TABLE 1
Cell-based optical detection methods, Methods Detection Probes Advantages Disadvantages
|
Methods |
Detection Probes |
Advantages |
Disadvanta |
|
Absorbance
Fluorescence Intensity
Luminescence
FRET |
X-gal (b-galactosidase substrate) Horseradish peroxidase
Fluo-3 (Ca indicator) Autofluorescence Fluorescein di-b-Dgalactopyranoside (bgalactosidase substrate) Cy3 and Cy5 Green fluorescent protein chimeras
Luciferin
Coumarin-fluorescein-based FRET substrate (blactamase), Cameleons |
Simple methodology
Easily used with antibodies
Bright signal, good for miniaturization Easily used with antibodies
Can be used for subcellular localization
Very sensitive due to low background
Ratiometric output, detects proteinprotein interactions and good for miniaturization |
Limited prospects for miniaturization
Autofluorescence
Interference by compounds
Low proton flux requires longer read out times
Sensitive to placement of probe and significant probe development |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
Conclusions
Most of the discussion in this commentary has focused on the generation and examination of small organic molecules, but obviously combinatorial libraries of peptides, nucleic acids, and oligosaccharides continue to be generated. The inherently higher molecular weight peptides, nucleic acids, and oligosaccharides can be evaluated with most of the in vitro assays, but cell-based assays are more problematic because of cellular entry barriers and susceptibility to catabolic processes. Expansion in the number, diversity, and general availability of small molecule, combinatorial chemical libraries seems likely to make these essential reagents for future pharmacological studies.
References
Boger DL, Tarby CM, Myers PL and Caporale LH (1996) Generalized dipeptidomimetic template: Solution phase parallel synthesis of combinatorial libraries. J Am Chem Soc 118:2109–2110.
Choong IC and Ellman JA (1996) Solid-phase synthesis: Applications to combinatorial libraries. Annu Rev Med Chem 31:309–318.
Curran DP and Wipf P (1997) Combinatorial definitions. Chem Eng News 75:6–7.
Dolle RE and Nelson KH (1999) Comprehensive survey of combinatorial library synthesis. J Comb Chem 1:235–282.
Drews J (1996) Genomic sciences and the medicine of tomorrow. Nature Biotechnol 14:1516–1518.
Gallop MA, Barret RW, Dower WJ, Fodor SPA and Gordon EM (1994) Applications of combinatorial technologies to drug discovery. Background and peptide combinatorial libraries. J Med Chem 37:1233–1251.
Gayo LM and Suto MJ (1997) Ion-exchange resins for solution phase parallel synthesis of chemical libraries. Tetrahedron Lett 38:513–516.
Gold L and Alper J (1997) Keeping pace with genomics through combinatorial chemistry. Nature Biotechnol 15:297.
Gonzalez JE and Negulescu PA (1998) Intracellular detection assays for highthroughput screening. Curr Opin Biotechnol 9:624–631.
Gonza´ lez JE, Oades K, Leychkis Y, Harootunian A and Negulescu PA (1999) Cellbased assays and instrumentation for screening ion-channel targets. Drug Discovery Today 4:431–439.
Gordon EM, Barrett RW, Dower WJ, Fodor SPA and Gallop MA (1994) Applications of combinatorial technologies to drug discovery. Combinatorial organic synthesis, library screening strategies, and future directions. J Med Chem 37:1385–1401.
Gray NS, Wodicka L, Thunnissen A-M WH, Norman TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, LeClerc S, Meijer L, Kim S-H, Lockhart DJ and Schultz PG (1999) Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science (Wash DC) 281:533–538.
Hogan JC (1996) Directed combinatorial chemistry. Nature (Lond) 384:17–19.
Hogan JC (1997) Combinatorial chemistry in drug discovery. Nat Biotechnol 15:328–330.
Houston JH and Banks M (1997) The chemical-biological interface: Developments in automated and miniaturised screening technology. Curr Opin Biotechnol 8:734–740.
Jung G (1996) Combinatorial Peptide and Nonpeptide Libraries: A Handbook. Weinheim, New York.
Nefzi A, Ostresh JM and Houghten RA (1997) The current status of heterocyclic combinatorial libraries. Chem Rev 97:449–472.
Nestler HP, Bartlett PA and Still WC (1994) A general method for molecular tagging of encoded combinatorial chemistry libraries. J Org Chem 59:4723–4724.
Obrecht D and Villalgordo JM (1998) Solid-Supported Combinatorial and Parallel Synthesis of Small-Molecular-Weight Compound Libraries. Pergamon Press Ltd., Oxford, UK.
Parlow JJ, Naing W, South MS and Flynn DL (1997) In situ chemical tagging: Tetrafluorophthalic anhydride as a “sequestration enabling reagent” (SER) in the purification of solution-phase combinatorial libraries. Tetrahedron Lett 38:7959–7962.
Pearce LL, Gandley RE, Han W, Wasserloos K, Stitt M, Kanai AJ, McLaughlin MK, Pitt BR and Levitan ES (2000) Role of metallothionein in nitric oxide signaling as revealed by a green fluorescent fusion protein. Proc Natl Acad Sci USA 97:477–482.
Pirrung MC (1997) Spatially addressable combinatorial libraries. Chem Rev 97:473–488.
Rice RL, Rusnak JM, Yokokawa F, Yokokawa S, Messner DJ, Boynton AL, Wipf Pand Lazo JS (1997) A targeted library of small molecule, tyrosine and dual specificity phosphatase inhibitors derived from a rational core design and random side chain variation. Biochemistry 36:15965–15974.
Siegel MG, Hahn PJ, Dressman BA, Fritz JE, Grunwell JR and Kaldor SW (1997) Rapid purification of small molecule libraries by ion exchange chromatography. Tetrahedron Lett 38:3357–3360.
Studer A, Hadida S, Ferritto R, Kim S-Y, Jeger P, Wipf P and Curran DP (1997)
Fluorous synthesis: A fluorous-phase strategy for improving separation efficiency in organic synthesis. Science (Wash DC) 275:823–826.
Wipf P, Cunningham A, Rice RL and Lazo JS (1997) Combinatorial synthesis and biological evaluation of a library of small-molecule Ser/Thr-protein phosphatase inhibitors. Bioorg Med Chem 5:165–177.
Yan B (1998) Monitoring the progress and the yield of solid-phase organic reactions on resin supports. Acc Chem Res 31:621–630.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org








