

About Authors
Vivekanandan Kalaiselven, Shatrunajay Shukla, Nikita Mishra and Pawan Kumar
vivekarts@rediffmail.com
Abstract
Medical devices are being used in healthcare facilities for diagnosis, monitoring, prevention and treatment of an array of diseases. To ensure the safety of the medical devices being used in healthcare industry it is of utmost importance to closely monitoring the adverse events associated with the medical devices through a robust, sustainable and scaled surveillance. Materiovigilance Programme of India (MvPI) provides a reliable system to report adverse events associated with medical devices. Under MvPI, various modalities to report adverse events associated with medical devices have been developed. These modalities include an editable medical device adverse event reporting form, a toll-free helpline number and a field safety corrective action form to notify the regulatory authority and healthcare professionals on corrective actions or recall by the manufacturer. Due to the emergence of the Coronavirus disease 2019 (COVID-19) pandemic, one-page editable form has been developed to boost the adverse event reporting of Personal Protective Equipments (PPEs). MvPI also coordinates with healthcare facilities and medical device industries across the country for reporting the medical device-related adverse events. The collected scientific data is utilized to develop regulatory policies and enhance measures to ensure the quality of medical devices. All the healthcare workers are, therefore, encouraged to report adverse events to MvPI. This chapter aims to describe the systems, procedures and modalities available for the reporting of Medical Device Adverse Events (MDAEs) in India, in order to intensify the nature of reporting and creating an environment that encourages the public to perform MDAE reporting.
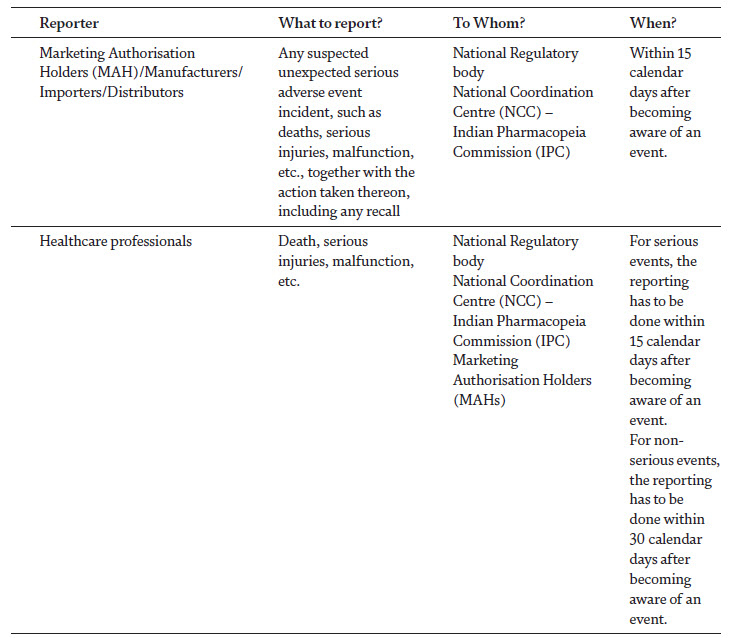
Introduction
Over the last years, medical devices have been playing a pivotal role in the diagnosis and management of a variety of diseases [1]. With the advancement in the technology and increased public demand for high quality medical care, the global medical device industry has surpassed USD 350 billion in annual revenue, and in India a growth rate of 20% has been seen in healthcare industry, while it is expected to reach USD 175 billion by 2020. These devices have also created substantial risks to the patients with high profile recalls [2]. Nearly 5,000 individual classes of medical devices, tens of thousands of medical device suppliers, and millions of healthcare providers exist worldwide, which clearly depicts that device-related issues are likely to occur. The outcome of an adverse event related to medical devices can be serious and result in illness, injury or even death, which have led experts to call for the monitoring of the safe and effective use of medical devices after its regulatory approval [3]. Materiovigilance Programme of India (MvPI) was launched in 2015 and has implemented a robust system to ensure the safety of medical devices. The aim of this programme is to identify the adverse events associated with the use of medical devices and to eliminate the device-related risks through a systematic reporting system [4]. In India, the Medical Devices Rules (MDR) were implemented in 2017 and became effective from January 1st, 2018. As per the MDR, G.S.R. 78 (E), published on January 31st, 2017, Chapter 4, Section 26 (ii) “the License Holder shall inform the State Licensing Authority (SLA) or Central Licensing Authority (CLA), as the case may be of the occurrence of any suspected unexpected serious adverse events and take necessary action thereon, including any recall within 15 days of such event coming to the notice of License Holder” [5] (Table 1). The MDR, in concurrence with MvPI, has significantly influenced the post marketing surveillance of medical devices among the healthcare professionals, by ensuring their quality and patient/user safety [5]. This chapter aims to describe the systems, procedures and modalities available for the reporting of Medical Device Adverse Events (MDAEs) in India, in order to intensify the nature of reporting and creating an environment that encourages the public to perform MDAE reporting.
MvPI Focus Groups for MDAE reporting
The following groups play a significant role in the smooth functioning of MvPI:
Medical Device Adverse Event Monitoring Centres (MDMCs) and Adverse Drug Reactions Monitoring Centres (AMCs)
Healthcare facilities, including district/government/private hospitals, and autonomous bodies are recognized as MDMCs and AMCs by the NCC-MvPI, IPC and NCC-PvPI, IPC respectively. The function of the MDMC is to raise awareness about the programme and reporting of MDAEs. The MvPI collects reports from the MDMCs, AMCs. Under the MvPI, 50 MDMCs have been identified so far in India to collect the report of the events associated with the use of medical devices [6]. The AMCs established under the Pharmacovigilance Programme of India (PvPI) are also participating in MDAE reporting. Around 311 centres have been identified in the country to report the adverse events resulting from the use of drugs/medical products [7].
Medical device industries
Medical device industries, including manufacturers, importers, distributors, etc., are approached and encouraged to report the MDAE, particularly with their own medical devices. As there may be chances of re-occurrences of the adverse events, medical device industries play a key role in medical device safety surveillance [8].
Healthcare professionals
Healthcare facilities include healthcare professionals, such as clinical specialists, biomedical engineers, nurses, pharmacists, hospital technology managers, and technicians, as well as patients. Healthcare professionals are in direct contact both with the patients and the medical devices used in the healthcare facilities, and are hence the key personnel in MDAE reporting [9].
Accreditation bodies
Accreditation bodies essentially identify the capability of the hospitals to deliver quality care. Indian healthcare institutions get accreditation from bodies such as the National Accreditation Board for Hospitals and Healthcare providers (NABH). The IPC has signed a Memorandum of Understanding (MoU) with the NABH, under the Quality Council of India (QCI), to ensure for total cooperation of the hospitals on the reporting of adverse events associated with medical devices in the hospital [10].
Modalities for MDAE reporting
The NCC for MvPI, IPC has developed the below-mentioned reporting tools to collect MDAEs. All the reporting tools are available on the IPC website. The healthcare professionals, MAHs and all the Personal Protective Equipments (PPEs) users are encouraged to report adverse events associated with medical devices [11].
MDAE reporting form
The MDAE reporting form primarily aims to collect the adverse events associated with the use of medical devices, In-Vitro Diagnostics (IVDs), and medical equipments. The healthcare professionals and others including, but not limited to, manufacturers, importers, distributors, and hospital managers are solicited to report the adverse events for known, unknown, serious, non-serious, frequent or rare adverse events. The MDAE reporting form assembles adverse event information associated with medical devices and consists of the following nine sections (Figure 1) [11].
General information
This section includes the date of report, i.e. the date in which the report was filled, and the type of report that specifies whether the event is Initial/Follow-up/Final/Trend. The initial report is the first event notification report which may include the minimal required information, for instance. Device information, details of adverse event and reporter details. A report may be marked as a follow-up report when additional information is available from the previously reported event. The report may be submitted as a final report when all the information associated with the event is available and collected. If the reporter is observing a significant number of similar adverse events, the reporter may tick the trend option [12].
Reporter details
This section comprises the type of reporter, along with the details of the reporter, including. Name, address, contact number and e-mail address. A reporter may be a manufacturer, importer, distributor, healthcare professional, patient, or other. The information provided in this section is kept confidential and only utilised for further follow-up.
Device category
This section refers to the general information about the medical device used. The device category section in the MDAE reporting form consists of three subsections, namely medical device, medical equipment and IVDs. The medical equipment and IVDs subsections refer to specific categories of medical devices. Medical devices requiring calibration, maintenance, repair, user training and decommissioning – are usually managed by clinical engineers. Medical equipment is used for the specific purposes of diagnosis and treatment of disease or rehabilitation following disease or injury. It can be used either alone or in combination with any accessory, consumable or other piece of medical equipment. Medical equipment excludes implantable, disposable or single-use medical devices. The IVD medical devices includes a medical device, used either alone or in combination, intended by the manufacturer for the in-vitro examination of specimens derived from the human body solely or principally to provide information for diagnostic, monitoring or compatibility purposes. All the other devices not covered under the definitions of medical equipments and IVDs should be included into the medical device subsection.
Device description
This section describes the specific details of the suspected medical device: device name or the brand name used for marketing of the device, manufacturing or import firm name and address, the information of local distributer, the lot/batch number, serial number, year of manufacturing. Furthermore, in case of medical equipment, the additional information also includes installation date, last calibration date and preventive maintenance date, and software version is also asked. Many countries use different nomenclature systems for naming the medical device. The most prominent codes known are the Global Medical Device Nomenclature (GMDN) and Universal Medical Device Nomenclature System (UMDNS). In the reporting form, the reporter has an option to add the nomenclature code of the device while reporting the event.

Figure 1. Pictorial representation of medical device adverse event (MDAE) reporting form.
Event description
This section includes the most important dates associated with the adverse event, such as the date in which the event or any near miss incident occurred, etc. Furthermore, this section also comprises the information about device operator, device location and the detailed description of the event. The reporter may mark an event as serious in case it fulfils the seriousness criteria described in MDR 2017 [5]. Otherwise, the event may be marked as non-serious.
Patient information
This section contains the patient information, including its medical history and final patient outcome after the adverse event has occurred. Additionally, the patient hospital ID, age, gender is also comprised in this section.
Healthcare facility information
This section includes the details of the hospital in which the event took place, as well as the details of a contact person at the hospital, for the further follow-up communication related to the adverse event.
Causality assessment
This section aims to collect the information regarding the investigation process carried out by the clinical specialists from the healthcare facility, or by the concerned personnel from the manufacturing organization, to further drawing out the root cause of the problem and the immediate action taken to reverse the adverse effect, if possible. The root cause will ascertain the most likely reason for the occurrence of the adverse event.
Manufacturer/Authorized representative investigation & action taken
This section provides the information related to the investigation methods performed by the manufacturer/authorized representative and device history, which includes a review of similar events occurred from the same batch/lot, the analysis report of the event related to the medical device, and the corrective/ preventive action/recall taken to prevent the patient from being affected by the device, if any. The MDAE form is designed in such a manner that it collects the maximum information required, which may be helpful for the identification of the MDAE and for creating the database of the medical device-related errors, thus enabling to trace the trend of adverse events associated with medical devices. The MDAE form may also help the medical device stakeholders to provide appropriate information and enhance the quality of the information collected.
PPE Reporting Form
During the prevailing situation regarding the COVID-19 pandemic, the NCC-MvPI specially designed a one-page editable MDAE reporting form, which primarily aims to collect the adverse events associated with the use of PPEs used for medical purposes (Figure 2) [11]. The information required to be filled in the reporting form under the different categories is as follows [11] :

Figure 2. Pictorial representation of personal protective equipment (PPE) reporting form.
General information
This section includes the exact date in which the event was reported to the NCC-MvPI, IPC, and the type of report that specifies whether the event is Initial/Follow-up. The initial report is the first notification about an adverse event submitted to the NCC-MvPI, IPC, once the reporter became aware of it. The follow-up report comprises the additional information about the previous report.
Reporter details
This section comprises the details of the reporter, including name, address, contact number and e-mail address. The information provided in this section is kept confidential and only utilized for the follow-up.
PPE type This section encompasses the type of PPE involved in the adverse event/reactiongloves, coverall, goggles, N-95 masks, shoe covers, face shields, body bags, triple layer medical mask, among others.
PPE details
This section describes the specific details of the PPE involved in the adverse event. These details include the brand name, manufacturer/importer/distributer name, batch number, model number, license number, unique certification code, test standard followed, manufacturing date and expiry date.
Location of event
This section refers to the location where the adverse event has occurred, and includes inpatient department, quarantine facilities, emergency department, etc.
Type of event This section comprises the seriousness of the event. If the event involves the following outcomes: death/life threatening/disability or permanent damage/ hospitalization/congenital anomaly, then it should be marked as serious. Otherwise, the event may be marked as non-serious.
User details
This section covers the details of the PPE user, including user initials, age, gender, etc.
Event description
This section includes the detailed description of an adverse event associated with PPEs.
Hospital/quarantine facility details
This section provides the details related to the hospital/quarantine facilities, including name, address and contact person. The PPE form is designed in such a manner that it collects all the required information, which may be used for the identification of PPE-related adverse events and for creating the database of such adverse events.
Field safety corrective action (FSCA) notification form
The FSCA [11] refers to any action taken to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device, including the: (i) device returned to the manufacturer, (ii) device design changes, (iii) device software upgrade, (iv) labelling changes, (v) changes in instructions for use or directions for use or technical manual, (vi) device destruction and (vii) device exchange. For more information, see ([13]).
Legal obligation
The submitted MDAE report does not have any legal implication concerning the reporters. The patients’ identity will be held under strict confidence and protected to its full extent. As the reporting programme is voluntary in nature, healthcare providers are encouraged to report adverse events for a better understanding of the risk associated with the use of medical devices, and to safeguard the health of the Indian population [14].
Essential data for effective reporting
This section includes the following information: date of event, reporter contact information, device name, manufacturer/importer/distributor details, catalogue number., lot/batch number., serial number., model number., date of implantation/explantation (if applicable), seriousness of the event, event description, patient history, patient outcome, healthcare facility information, root cause and corrective/preventive action [13].
Factors contributing to a serious adverse event
The improper functioning of the medical devices, manufacturing defects, design and labelling issues, user and procedural errors are some examples of the major contributing factors that can lead to the occurrence of a serious adverse event if underestimated [15].
Helpline facility for reporting adverse events
The IPC has already launched a toll-free helpline facility, helpline Number- 1800 180 3024 (Monday to Friday- 9:00 am to 5:30 pm), for the reporting of adverse drug reactions by healthcare professionals and others [16]. Currently, this facility is also being extended to the report of any adverse event associated with the use of medical devices. Both the data management and the procedure adopted to receive the information from the healthcare professionals, patients and others are given in Figure 3.
Enrolment process as MDMC under MvPI
Healthcare facilities including district/government/private hospitals, and autonomous bodies are recognized as MDMCs and AMCs respectively by the NCC-MvPI, IPC and NCC-PvPI, IPC. The function of MDMC is to raise awareness about the programme and reporting of MDAEs. A ‘Letter of Intent’ is required to be submitted by the head of the Institution/hospital for participating in this nationwide programme to monitor MDAEs [17]. After the suitability examination by the competent authority, the proposed centre may be recognized as an MDMC under MvPI. These centres are expected to collate data on adverse events associated with medical devices and IVDs under the MvPI and report them to the NCC-MvPI, IPC. For the proper functioning of MvPI activities in their respective centres, a research associate will be appointed by the NCC-MvPI, IPC [17]. The research associate will be responsible for the collection of reports and conduction of training programs on materiovigilance, aiming to sensitize the healthcare professionals and the general public. The technical team at the MDMC performs the validation of the report by carrying out the causality assessment to identify any causal/temporal relationship between the event and the medical device.
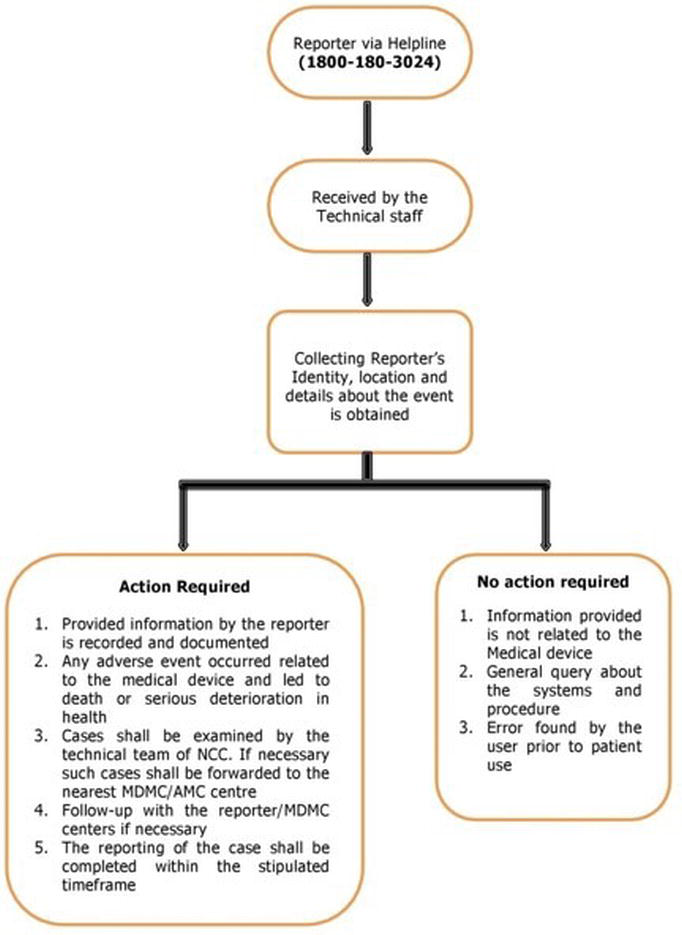
Figure 3. Flow diagram representing the report of adverse events related to medical devices through helpline.
The workflow for determining the report responsible for the identification of adverse events significantly related to medical devices is shown at Figure 4.
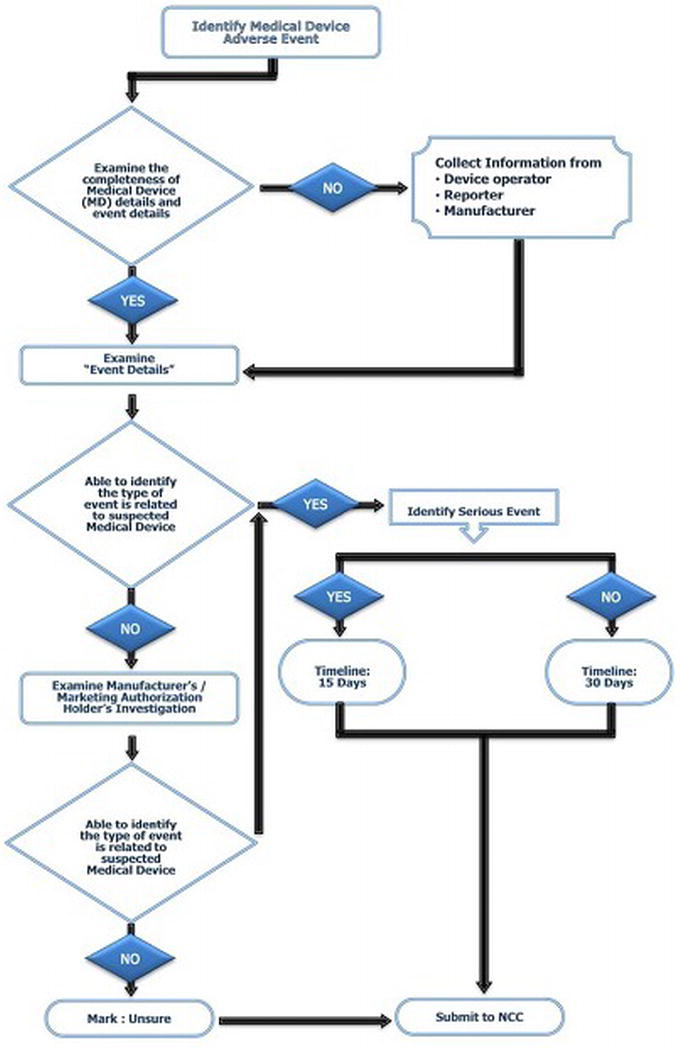
Figure 4. Medical device-related adverse events identification flowchart used at medical device adverse event monitoring centres (MDMC)
Report Handling and Management
Initially, the reports are collected and analysed at the NCC-MvPI, IPC, by applying the globally recognized scientific standards/parameters to ensure the quality of the reports. In the second level, these analysed cases are forwarded to the subject expert group panel for review, and technical interpretation is drawn considering both the clinical, as well as technical aspects. In the third level, these reports are placed before the core technical committee for the conclusions and recommendations, and are further forwarded to the national regulatory authority for implementation of the necessary actions (Figure 4) [19].
Data Generated
The NCC has collected the reported adverse events and provided a comparison of the serious adverse events reported in the index period from January to December during the years of 2018, 2019 and 2020. In total, NCC has received 3187 adverse events, consisting of 1986 serious and 1201 non-serious events. Out of the serious adverse events reported, 23% were reported in 2018, 37% in 2019 and 40% in 2020. When comparing the reported data, an increase of 75% in serious adverse event reporting could be observed. Out of the adverse events reported, 73% of the reports were received from MAHs, 23% from MDMCs and 4% from AMCs [19]. This confirms and highlights the importance of the modalities developed, as they have significantly helped to improve the reporting of adverse events related to medical devices.
Conclusion
The tools developed for reporting may stimulate the communication between medical device users and the regulatory authorities to closely monitor medical device safety. In order to generate proper regulatory decisions and to ensure the quality and efficacy of the medical devices that are being sold and distributed in the Indian market, MvPI has shown to provide a robust and sustainable system for collecting and reporting adverse events associated with medical devices. This will highly encourage all the healthcare professionals, MAH and the public to efficiently report adverse events associated with medical devices.
References
[1] Materiovigilance and Medical Devices, doi.org/10.1007/978-3-319- 07653-9_21 [Online]. Available: https:// link.springer.com/chapter/10.1007/ 978-3-319-07653-9_21 (accessed Mar. 15, 2021).
[2] Medical Device Recalls, FDA, Sep. 09, 2020. [Online]. Available: https:// www.fda.gov/medical-devices/medicaldevice- safety/medical-device-recalls (accessed Feb. 24, 2021).
[3] Analysis of FDA-Approved Orthopaedic Devices and Their Recalls, JBJS, vol. 98, no. 6, pp. 517-524, Mar. 2016, doi: 10.2106/JBJS.15.00286 [Online]. Available: https://pubmed. ncbi.nlm.nih.gov/26984921/ (accessed Feb. 24, 2021).
[4] Materiovigilance programme of India (MvPI): A step towards patient safety for medical devices. [Online]. Available: https://www.researchgate.net/ publication/338065787_MATERIO VIGILANCE_PROGRAMME_OF_ INDIA_MvPI_A_STEP_TOWARDS_ PATIENT_SAFETY_FOR_MEDICAL_ DEVICES (accessed Feb. 24, 2021).
[5] Medical device rules 2017, [Online]. Available: https://cdsco.gov.in/opencms/ resources/UploadCDSCOWeb/2018/ UploadGazette_NotificationsFiles/ MedicalDeviceRulegsr78E.pdf (accessed Feb. 24, 2021).
[6] Materiovigilance: Current status in India analogous to its global status, DOI:10.5281/zenodo.4299028 [Online]. Available: https://zenodo.org/ record/4299028#.YE9F0NIzZ0w.html (accessed Mar. 15, 2021).
[7] Pharmacovigilance Programme of India (PvPI) Updates - Indian Pharmacopoeia Commission, [Online]. Available: https://ipc.gov.in/mandates/ pvpi/pvpi-updates.html (accessed Feb. 24, 2021).
[8] Implementation of adverse event reporting for medical devices, India, [Online]. Available: https://www. researchgate.net/publication/ 339488352_Implementation_of_ adverse_event_reporting_for_medical_ devices_India (accessed Feb. 24, 2021).
[9] MATERIOVIGILANCE: AN EMERGING DISCIPLINE [Online]. Available: https://www.worldwide journals.com/international-journal-ofscientific- research (IJSR)/fileview.php? val=June_2018_1527932925__36.pdf (accessed Mar. 15, 2021).
[10] Pivotal role of Pharmacovigilance Programme of India in containment of antimicrobial resistance in India, doi: 10.4103/picr.PICR_29_18 [Online]. Available: https://www.ncbi.nlm.nih. gov/pmc/articles/PMC6647896/ (accessed Feb. 24, 2021).
[11] Medical Devices Adverse Event Reporting Tools - Indian Pharmacopoeia Commission [Online]. Available: https://ipc.gov.in/mandates/pvpi/ materiovigilance-programme-of-indiamvpi. html?id=597:medical-devicesadverse- event-reporting-tools&catid=2 (accessed Feb. 24, 2021).
[12] Adverse events reporting of medical devices, HSA. [Online]. Available: https://www.hsa.gov.sg/medicaldevices/ adverse-events (accessed Feb. 24, 2021).
[13] Guidance Document: Materiovigilance Programme of India (version 1.2). [Online]. Available: https://ipc.gov.in/images/ Guideline_-_11-03-2020.pdf (accessed Feb. 24, 2021).
[14] Materiovigilance: An Indian perspective, Perspect Clin Res, vol. 9, no. 4, pp. 175-178, 2018, doi: 10.4103/ picr.PICR_26_18. [Online]. Available: https://pubmed.ncbi.nlm.nih. gov/30319948/ (accessed Feb. 24, 2021).
[15] Medical Device Reporting (MDR): How to Report Medical Device Problems, FDA, Oct. 05, 2020. https:// www.fda.gov/medical-devices/medicaldevice- safety/medical-devicereporting- mdr-how-report-medicaldevice- problems (accessed Feb. 24, 2021).
[16] V. Kalaiselvan, P. Mishra, and G. N. Singh, “Helpline facility to assist reporting of adverse drug reactions in India,” WHO South East Asia J Public Health, vol. 3, no. 2, p. 194, Jun. 2014, doi: 10.4103/2224-3151.206737[Online]. Available: https://pubmed.ncbi.nlm.nih. gov/28607307/(accessed Feb. 24, 2021).
[17] Letter of Intent for enrolling as medical device adverse event monitoring center under materiovigilance programme. [Online]. Available: https://ipc.gov.in/images/ pdf/File715.pdf (accessed Feb. 24, 2021).
[18] Materiovigilance Programme of India, DOI: 10.5958/0974-360X. 2021.00204.3 – An Overview. [Online]. Available: https://rjptonline.org/ HTMLPaper.aspx?Journal=Research%20 Journal%20of%20Pharmacy%20 and%20Technology; PID=2021-14-2- 100 (accessed Mar. 15, 2021).
[19] S. Shukla et al., “Implementation of adverse event reporting for medical devices, India,” Bull World Health Organ, vol. 98, no. 3, pp. 206-211, Mar. 2020, doi: 10.2471/BLT.19.232785. [Online]. Available: https://apps.who. int/iris/handle/10665/331375 (accessed Feb. 24, 2021).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT admin@pharmatutor.org
FIND OUT MORE ARTICLES AT OUR DATABASE








