

About Authors
MANISHA KOTADIYA*, JAYDEEP SAVALIYA
*Shree Swaminarayan Sanskar Collage of Pharmacy Zundal, Gandhinagar, Gujarat,
Email Id: manishakotadiya3@gmail.com,
FTF Pharma Pvt. Ltd. – Ahmedabad, Email ID: jaydeepsavaliya27@gmail.com
DOWNLOAD AS PDF
ABSTRACT
Aromatase is an enzyme complex which is assigned in the last step of estrogen biosynthesis; it increases total estrogen levels by converting androgens to estrogens in tissues like breast, ovary, testis, placenta, brain and adipose. Estrogen causes increased proliferation of tumor cells. It has been shown that the increased expression of aromatase is responsible for high estrogen levels in breast cancer. Therefore, in the treatment, aromatase inhibitors are being tested. The purpose of this study is to demonstrate the effects of aromatase inhibitors on breast health by their mechanisms, to compare their effectiveness according to the other agents in the treatment of breast cancer. For this purpose, literature findings about this subject were summarized. In addition, the resistance mechanisms that can develop against the therapy and the ongoing studies about new treatment strategies for preventing the development of resistance were also mentioned. As a consequence it was understood that the reduction of estrogen synthesis through aromatase inhibition provide greater therapeutic benefits with fewer side effects. These days, aromatase inhibitors are the best option especially for the treatment of postmenopausal women’s breast cancer. Keywords: Aromatase, Estrogen, Breast Cancer, Resistance.
I. INTRODUCTION
Cancer is the second most common cause of death after cardiovascular diseases. Each year, 7 million people die from cancer, and 11 million new cases are diagnosed worldwide. By 2050, the global burden is expected to grow to 27 million new cancer cases and 17.5 million cancer deaths simply due to the growth and aging of the population.1 The prevalence of different tumors varies worldwide; lung, colorectal, prostate and breast cancers account for more than fifty percent of all cancers in Europe, where as gastric and esophageal malignancies predominates in far East. This shows the importance of environmental factors upon occurrence of cancer. Other risk factors are related to life style such as smoking (lung cancers) or related to the work place, such as exposure to dye chemicals (bladder cancers).2
Cancer develops when a cell escapes from these normal regulatory mechanisms and proliferates uncontrollably, acquiring the ability to invade surrounding normal tissues and metastasis to distant sites. The branch of medicine concerned with the study, diagnosis, treatment and prevention of cancer is oncology.3
Tumors of cancer are classified as
a. Benign tumors a benign tumor is composed of cells that will not invade other unrelated tissues or organs of the body, although it may continue grow in size abnormally. These are localized tumors, which have a limited potential and do not usually produce serious side effects. Examples are Adenoma, Fibroma.
b. Malignant tumor a malignant tumor is composed of cells that invade the basement membrane and invade or spread to other parts of the body. This occurs either by direct extension to neighboring organs and/or tissues or by metastasizing to distant sites by means of vascular systems, the lymphatic system, or by seeding or implantation of cancer cells in body cavities. Types of malignant tumors are
1. Carcinoma, it is malignant tumor arising from epithelial tissue. Examples are Adenocarcinoma, Papillary carcinoma.
2. Sarcoma, it is malignant tumor arising from mesenchyme tissue. Examples are Myxosarcoma.
3. Sarcoma.4,5
Causes of Cancer
The main causes of cancer are discussed under the following categories.6-7
A. Radiation: High levels of radiation like those from radiation therapies and X-rays (repeated exposure) can damage normal cells and increase the risk of developing leukaemia, as well as cancers of the breast, thyroid, lung, stomach and other organs. UV radiation from the sun is directly linked to melanoma and other forms of skin cancer.
B. Viruses: Some viruses, including hepatitis B and C, human papilloma viruses (HPV) and the Epstein Barr virus which causes infectious mononucleosis, have been associated with increased cancer risk. Immune system diseases, such as AIDS, can make one more susceptible to some cancers.
C. Chemicals: Long term exposure to chemicals such as pesticides, uranium, nickel, asbestos, radon and benzene can increase the risk of cancer. Such carcinogens may act alone or in combination with another carcinogen, such as cigarette smoke, to increase the risk of cancer and other lung diseases.
D. Tobacco: Cigarette smoking and regular exposure to tobacco smoke greatly increase risk to lung cancer. Cigarette smokers are more likely to develop several other types of cancers like those of the mouth, larynx, oesophagus, pancreas, bladder, kidney and cervix.
E. Alcohol: Heavy drinkers face an increased risk of cancers of the mouth, throat, oesophagus, larynx and liver. Some studies suggest that even moderate drinking may slightly increase the risk of breast cancer.
F. Diet High-fat and high cholesterol diets are proven risk factors for several types of cancers such as those of the colon, uterus and prostate. Obesity may be linked to breast cancer among older women as well as to cancers of the prostate, pancreas, uterus, colon and ovary.
G. Hereditary: Risk Factors Twenty percent of cancers are hereditary. This means that the abnormal gene responsible for causing cancer is passed from parent to child, posing a greater risk for that type of cancer in all descendants of the family.
H. Genetics: A theory exists with some scientific support, that certain smokers have a higher risk of smoking-induced lung cancer than others because of their genetic make-up.
I. Family History: Many cancers are associated with having a family history of cancer. Breast, ovarian, prostate and colon are some of these cancers.
Breast Cancer :
Breast cancer is a malignant growth that begins in the tissues of the breast generally originating from breast tissue, most commonly from the inner lining of milk ducts or the lobules that supply the ducts with milk. Breast cancer is one of the most prevalent cancers in the world today due to its high incidence and about more than 1.1 million women worldwide is newly diagnosed with breast cancer annually. The most common types of breast cancer are ductal carcinoma (85-90% of all cases) and lobular carcinoma (8% of all cases). Less common are inflammatory breast cancer and Paget’s disease of the nipple.8 worldwide, breast cancer is the most common cancer in women after skin cancer, representing 16% of all female cancers. The rate is more than twice that of colorectal cancer and cervical cancer, and about three times that of lung cancer. Mortality worldwide is 25% greater than that of lung cancer in women. In 2009, breast cancer caused 464,000 deaths worldwide (7% of cancer deaths).9
Approaches used in the Treatment of Breast Cancer :
Two pharmacological approaches are currently used to reduce the effect of estrogen and to treat the breast cancer.
• The first approach involves the inhibition of action by antiestrogen, which interact with the estrogen receptor in antagonistic fashion.
• The second more elegant approach is inhibition of estrogen production using inhibitors enzyme aromatase.
Aromatase Enzyme:
Aromatase is protein and redox partner complex consist of two components (fig.1): aromatase cytochrome P450 (CYP 450-arom), a flavoprotein, NADPH-cytochrome P450 reductase. It is a member of the cytochrome P450 19 super family of enzymes and shares a number of common structural features common to all cytochrome P450 species. 15 Most notably, toward the carboxy terminus there is the heme-binding region containing a totally conserved cysteine residue that serves as the fifth coordinating ligand of the heme iron. Instead of the nitrogenous base in other b-type cytochromes, a thiolate ion is present at this site which is responsible for catalytic properties of this family of heme proteins. Upstream of this domain is a region of over 20 amino acids that is highly conserved in all aromatase species, and upstream of this is another region of highly conservation, namely the portion of the I helix believed to form the substrate binding pocket proximal to the heme prosthetic group.16
Structure of Aromatase :
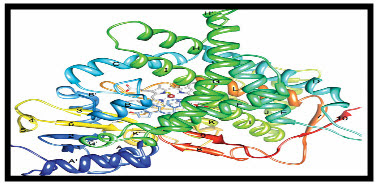
Figure 1 : A ribbon diagram of aromatase. The N terminus, starting at residue 45 is colored dark blue and the C terminus ending at residue 496 is colored red. The 5-helices are labelled from A to L and I-strands are numbered from 1 to 10. The heam group, the bound androstenedione molecule at the active site and its polar interactions are shown.
Mechanism of Aromatase Enzyme :
Conversion of androgens to estrogen by aromatase comprise of three sequential oxidation step, each one requiring 1 mole of
O 2 and NADPH (fig. 2). 18
Androgens + 3 NADPH + 3 H + + 3O2 ---------------> Estrogen + HCOOH + 3 H2O + 3 NADP +
Step-1: The Androgen is hydroxylated at C19 atom to afford the hydroxy intermediate by the classical enzyme mediated hydrogen atom abstraction hydroxyl radical rebound mechanism with the retention of configuration. 19-20
Step-2: 19 hydroxy steroid undergoes second hydroxylation of C19 atom to give C19 gem diol without an apparent kinetic isotope effect. This then undergoes rearrangement with a loss of 1 mole of water to the corresponding aldehyde. 21
Step-3: Oxidative Cleavage of C10-C19 bond resulting in aromatization of the steroid a ring and release of formic acid. 22, 23
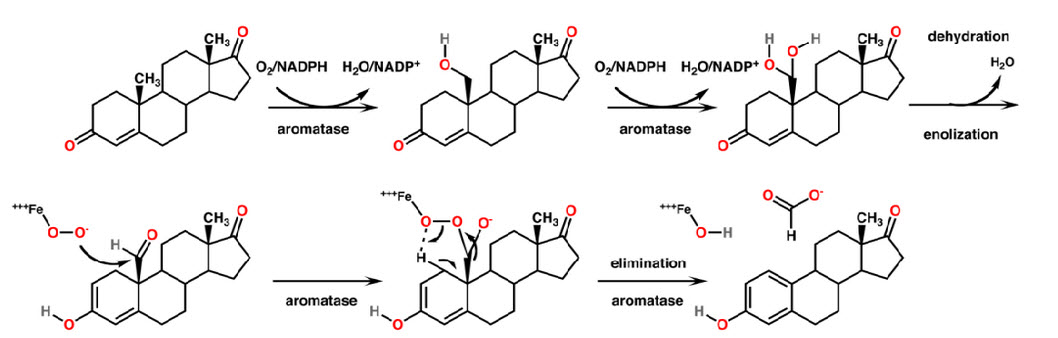
Figure 2 : Mechanism of Aromatase Inhibitors
Aromatase in Breast Cancer :
Expression of aromatase is highest in or near breast tumour sites. Several studies have reported that serum level of estrogen is very low in post-menopausal women but concentration of estrogen in breast tissues are 4-6 fold higher than serum level estrogen, concentration in tumour are even higher as compare to normal breast tissues.25 Tumour estrogen may be drive by two sources first by active uptake from circulation second from local aromatization of adrenal androgens, second source is more important suggested by isotope labelled estrogen studies. Further the increased expression of aromatase cytochrome P450arom observed in breast cancer tissues is also associated with a switching of predominant promoter PI.3 or PI.4 (normal breast tissue promoter9) to promoter PII.39 Promoter PII mainly present in ovary and breast cancer. Change in promoter also leads to change in the regulation of estrogen biosynthesis. Promoter PII regulated through cAMP mediated pathways. Prostaglandin E2 (PGE2) increases intracellular cAMP level and stimulates estrogen biosynthesis. With aromatase inhibitor therapy, there is a precipitous drop in the intratumoral concentrations of estradiol and estrone together with a corresponding loss of intratumoral aromatase activity.26
Aromatase Inhibitors :
Inhibitors of aromatase are divided into two main classes (Table.1).
Type 1 inhibitors interact with the substrate binding sites of the enzymes. These are structurally close to androgens and as a consequence they may have hormonal activity. In some cases inhibition may be mechanism-based in that the drugs when bound to the catalytic site of the enzyme are metabolized to intermediates, which attach irreversibly to the active site thereby blocking the activity. Such inhibitors are specific, only inhibiting molecules foe which they are substrates. 27-28
Type 2 inhibitors interact with the heme moiety of the CYP prosthetic group of the of the aromatase molecule. These inhibitors contain favorably positioned heteroatom’s usually in the imidazole and triazole ring that are capable of binding to CYP enzymes such that their heterotoms coordinate the heme iron. Type 2 inhibitors are reversible and the resultant estrogen blockade is dependent on continuous presence of the drug. 29
Table: 1: Types of steroidal and non-steroidal aromatase inhibitors.
|
Generation |
Type-I (Steroidal) inhibitors |
Type-II (Nonsteroidal) inhibitors |
|---|---|---|
|
First |
Testolectone |
Aminoglutethimide |
|
Second |
Formestane |
Fadrozole, Rogletimide |
|
Third |
Exemestane |
Vorozole, Anasatrozole, Letrozole |
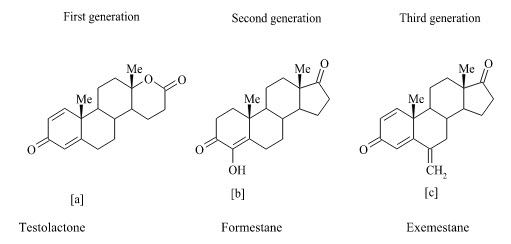
Type-I Steroidal inhibitors :
The steroidal group includes inhibitors that act in a competitive manner and certain others as enzyme inactivators or suicide inhibitors. Other terms that are used for these inhibitors are “enzyme-activated irreversible inhibitors,” “suicide substrates,” and “suicide inactivators”. Steroidal inhibitors are aromatase substrate analogs having shape similarity to androstenedione, which is a more hydrophobic substrate than others, and presents the best affinity for the enzyme. Testololactone (a) is used in breast cancer treatment for some 20 years and subsequently it has been reported to be a first generation aromatase inhibitor. Formestane (b) (4-hydroxyandrostenedione; 4-OHA) is a structural analog of androstenedione and is thus a highly specific aromatase inhibitor. It was the first steroidal suicide-type (Type l) aromatase inhibitor to enter clinical trials. The 4-OHA molecule binds irreversibly to the enzyme active site and acts as a suicide inhibitor with prolonged action. Exemestane (c) is another steroidal inactivator (mechanism-based) which has been approved more recently for clinical use and it appears to be more potent than 4-OHA.
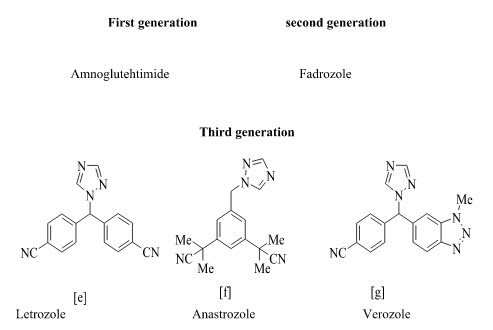
Type-II: Non-Steroidal Inhibitors :
Type II inhibition is usually reversible; estrogen blockage being dependent upon the continued presence of the drug. Non-steroidal inhibitors are mostly imidazole and triazole derivatives based on the structure of antifungal agents that inhibit P450 enzymes. Aminoglutethimide was the prototype nonsteroidal inhibitor of aromatase. Aminoglutethimide was the first inhibitor evaluated in clinical studies for treatment of hormone-dependent breast cancer. Santen et al extensively evaluated the kinetic and hormonal effects of aminoglutethimide in breast cancer and first proposed aromatase inhibition as the primary mechanism of action of aminoglutethimide therapy.32 The imidazole compound fadrozole is a potent, competitive inhibitor of aromatase, both in vitro and in vivo. Fadrozole, referred to as a “second generation inhibitor,” is more selective than aminoglutethimide and its inhibitory activity is 700 times more potent. The third-generation inhibitors possess a specificity that appears to be nearly complete in clinical doses with no effect on aldosterone, progesterone corticosterone biosynthesis. Recent clinical studies have shown that aromatase inhibitors, especially the third-generation, are more effective than a higher degree of specificity has been reached with the new generation of triazole derivatives (letrozole and anastrozole). These newer agents are 100-3000 times more active than aminoglutethimide and inhibit whole-body aromatization by a factor greater than 96%. Letrozole (Femara; Novartis, Basel, Switzerland) is a potent nonsteroidal aromatase inhibitor that produces approximately 99% inhibition of estrogen biosynthesis at a dose of 2.5 mg/d in patients. Anastrozole (Arimidex; Astra-Zeneca, London, UK) is a potent nonsteroidal aromatase inhibitor in patients, decreasing plasma estradiol levels in a dose-dependent manner.33 Vorozole is another third generation non-steroidal oral aromatase inhibitor which is highly potent and specific for aromatase. Its clinical efficacy appears to be similar to that of anastrozole and letrozole.
Other non-steroidal aromatse inhibitor
Apart from steroidal and nonsteroidal inhibitors, several flavones and isoflavones have also demonstrated aromatase inhibitory activity. However, these natural products demonstrate numerous biological activities and interact with various enzymes and receptor systems of pharmacological significance, thus limiting their therapeutic usefulness.
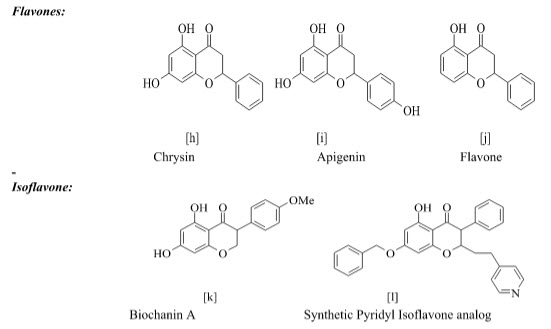
Table 2 : Comparison between steroidal and Non- steroidal Inhibitors
|
Type of inhibitor |
Advantages |
Disadvantages |
|
Non-Steroidal Inhibitors |
Less expected hormonal properties, Highly potent, Good pharmacokinetic Profile, Oral bioavailability. |
Less selective for enzyme, Musculoskeletal Problems, Shorter half-life. |
|
Steroidal Inhibitors |
More selective, Longer half-life (inactivation feature), Highly enzyme specific, Prolonged inhibition, Less toxic. |
Poor oral bioavailability, Androgenic effect on high dose, Metabolism by other CYP enzyme. |
II. AROMATASE INHIBITORS : LITERATURE REVIEW
A. Benzophenones
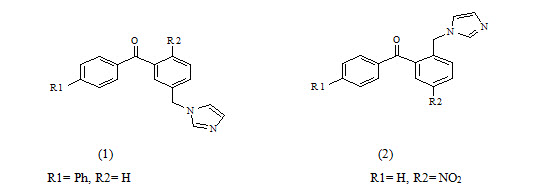
Silvia Gobbi et al have synthesized Imidazolylmethylbenzophenones and evaluated for aromatase Inhibitory activity. They found a new class of potent aromatase inhibitors endowed with high selectivity with respect to 17-hydroxylase/17, 20-lyase (CYP17). Compounds 2a and 2b proved to be the most potent inhibitors having IC50=7.3 and 5.3 respectively. Because of the substitution at phenyl ring to p-position give higher activity.
Kanamaru et al Physiologically active substance TAN-931 from Penicillium funiculosum and its derivatives as aromatase inhibitors The compound 3 in vitro inhibited aromatase from a homogenized human placenta microsome with IC50 of and 4.3 g/mL, resp.27
B. Flavones:
Kim et al reported the design, synthesis and biological evaluation of a novel series of 2-(4’-pyridylmethyl) thioisoflavones (4) as the first example of synthetic isoflavone-based aromatase inhibitors. The compounds were screened for humane placental microsomal aromatase assay. The compounds with following substitution were founds to be most active.

Recanatini et al reported the design and synthesis of chromone (5) and xanthone (6) derivatives as a new class of non-steroidal aromatase inhibitors. The compounds were screened for inhibition of the P450 enzymes aromatase and 17α-Hydroxylase/C17, 20-Lyase. The design of the new compounds was guided by a COMFA model.

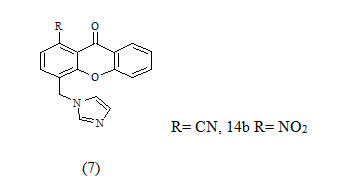
The xanthone derivatives 7a and 7b were found to be more potent with IC50 values 43 and 40 nm respectively, which exceeded the potency of the known reference drug fadrozole and showed good selectivity with respect to P450 17.because of this compound contain CN and NO2 its good electron withdrawing group shown give good activity Thus, they might be new leads for the development of drug candidates for androgen-dependent diseases.
Gobbi et al reported the synthesis of a Series of Flavone Derivatives as Potent Nonsteroidal Inhibitors of the Cytochrome P450 Aromatase Enzyme. The compounds 8a-f shows activity in Nano molar range. These modifications and the removal of the 7-methoxy group led to compounds showing inhibitory activity in the Nano molar range comparable to the marketed drug fadrozole.
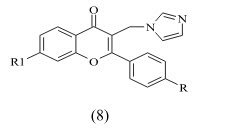
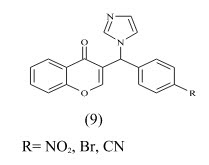
Cavalli et al synthesized enantioselective non-steroidal aromatase inhibitors through multidisciplinary medicinal chemistry approach and have done 3D QSAR study. The derivative with nitro substitution is most active with IC50 value 2.3± 1.1 nm.
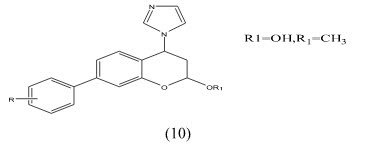
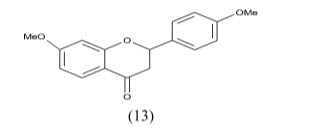
Chuliya et al Synthesis and biological evaluation of (10) 4-imidazolylflavans as nonsteroidal aromatase inhibitors. Among these molecules, 4-hydroxy-4-imidazolyl-7-methoxyflavan is only 2.2-fold less active than the letrozole IC50=41nM.a 7-hydroxyl group is essential for enhanced aromatase inhibitory activity while a 7-methoxy group is also responsible for an anti-aromatase effect. Finally, we demonstrated that additional hydroxyl groups at position 3- and/or 40 on the 7-methoxyflavanone skeleton led to an increase in aromatase inhibition.
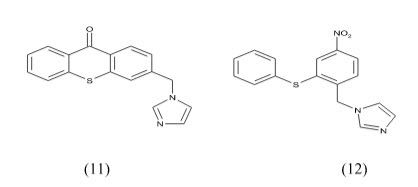
Hartmann et al Synthesis, Biological Evaluation and Structure-Activity Relationships Investigation A new series of molecules were designed and synthesized, in search for potent and selective aromatase inhibitors. The substitution of the heterocyclic oxygen atom in the xanthone core by a sulfur atom and/or increase in structure flexibility seemed to be favorable for the interaction with the enzyme. Among the new compounds, I and II (IC50 3.98 and 5.59 nM, respectively.) resulted the most potent aromatase inhibitors synthesized in this project.
Maiti, Arup et al Synthesis and Biological Evaluation of (±) Abyssinone II and Its Analogues as Aromatase Inhibitors for Chemoprevention of Breast Cancer. An efficient and economical synthesis of the naturally occurring aromatase inhibitor Abyssinone II was performed. As a result of these structural changes, the most active flavone of the series (I) was 20 times more potent than (±)-Abyssinone II (IC50 40.95 µM).
Mohammed, et al A Facile synthesis of Chrysin-derivatives with promising activities as aromatase inhibitors. Flavones such as Chrysin show structural similarities to androgens, the substrates of human aromatase, which converts androgens to estrogens. Aromatase is a key target in the treatment of hormone-dependent tumors, including breast cancer 35
C. Pyridine:
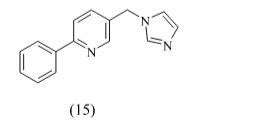
Hille et al developed a series of highly active and selective inhibitors of CYP11B1 for the treatment of cortisol-dependent diseases and also tested against steroid-genic CYP enzymes. Compound 15 (IC50 = 152 nM) is the first CYP11B1 inhibitor showing a rather good selectivity toward the most important steroid-genic CYP enzymes aldosterone synthase (CYP11B2), the androgen-forming CYP17, and aromatase (estrogen synthase, CYP19).because of this compound contain pyridine is spacer group give better active than other.
D. Indenodiazine
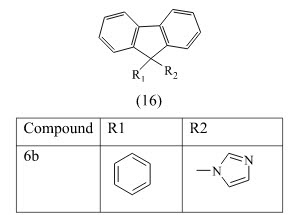
Leonetti et al have designed and synthesized a library of compounds bearing an imidazole or triazole ring linked to an indenodiazine and coumarin. The compounds were screened for aromatase inhibition. The most active compound is derivative 16b with an IC50 74 nM and a good CYP19/CYP17 selectivity, because of this compound contain imidazole which is bind to heme so it give good active than other.
E. Tetrahydroindolizin and Indanone
Sonnet et al reported design and synthesis of 3-phenyl-2-(pyridin-4-ylmethylen) indan-l-one (7) and 6-(4-chioro-phenyl)-7-(pyridin-4-ylmethylene)-5, 6, 7, 8-tetrahydroindolizin-8-one (8) and evaluated for aromatase inhibitors. The compounds 17 and1 8 have IC50 value 0.2 µM and 0.8 µM respectively. As these compounds contain 4-chlorophenyl its bind to serine residue give good activity
CONCLUSIONS
With the progress in medicinal chemistry of aromatase inhibitors, we have different strategies for inhibition of aromatase and different classes of chemical compounds to treat estrogen dependent diseases. Availability of large number and different classes of aromatase inhibitors aided to design optimal pharmacophore pattern to inhibit aromatase. The three compounds of interest anastrozole, letrozole and Exemestane, all have comfortable pharmacokinetic profile with lengthy half-lives allowing once daily administration without significant drug interactions. The third generation aromatase inhibitors are both remarkably potent and specific endocrine agents inhibiting aromatase activity. Their therapeutic potential consequently is much greater than the earlier prototype drugs. Aromatase inhibitors have also been used in the neoadjuvant setting, where they have been shown to achieve higher response rates than tamoxifen and decrease the need of major surgery. Finally, though third generation aromatase inhibitors are emerging as potential endocrine agents for the treatment of breast cancer, there is plenty of scope and need for improvement.
REFERENCES
1. Cancer Facts & Figures 2009. American Cancer Society Center in Atlanta, Georgia: Inc. 1-72.
2. Global Cancer Facts & Figures 2007. American Cancer Society Center in Atlanta, Georgia: Inc. 1-59.
3. Kenneth KW, Vogelstein B, 2002. Introduction, in: The Genetic Basis of Human Cancer, 2nd, illustrated revised Edn, McGraw-Hill Medical Pub Division, New York, 5.
4. National Cancer Institute; National Cancer Institute (NCI), United States Federal government's National Institutes of Health Bethesda, MD, “Common Cancers topics”, http://www.cancer.gov/cancertopics/wyntk/cancer/page4.
5. Merck Manua Online, Breast Cancer, http://www.merck.com/mmpe/print/sec18/ch253/ch253e.html.
6. American Cancer Society. Breast Cancer Facts & Figures 2009. Atlanta: American Cancer Society, Inc. 1-40.
7. Kellis J.T and Vickery L.E, 1987. Purification and Characterization of Human Placental Aromatase Cytochrome P-450. J. Biol. Chem. 262, 4413-4420.
8. Graham-Lorence S, Khalil M.W, Lorence M.C, Mendelson C.R, Simpson E.R 1991.Structure-function relationships of Human Aromatase Cytochrome P-450 using Molecular modeling and Site-directed mutagenesis. J. Biol .Chem. 266, 11939-11946.
9. Ghosh D, Griswold J, Erman M, Pangborn W. 2009. Structural basis for Androgen specificity and Estrogen synthesis in Human Aromatase. Nature. 457, 219-231.
10. Thompson E.A, Siiteri P.K. 1974. Utilization of Oxygen and Reduced Nicotinamide Adenine Dinucleotide Phosphate by Human Placental Microsome during Aromatization of Androstenedione. J. bio. Chem. 249, 5364-5372.
11. Miyairi S, Fishman J. 1983. Novel method of evaluating biological 19-Hydroxylation and Aromatization of Androgens. Biochem. Biophys Res Commun. 117, 392-398.
12. Miyairi S, Fishman J. 1985. Radiometric analysis of oxidative reactions in Aromatization by Placental microsome. Presence of differential isotope effects. J. Biol. Chem. 260, 320-325.
13. Osawa Y, Shibata K, Rohrer D, Weeks C, Duax W.L. 1975. Reassignment of the absolute configuration of 19-substituted 19-hydroxysteroids and Stereo mechanism of Estrogen Biosynthesis. J. Am Chem. Soc. 97, 4400-4402.
14. Akhtar M, Calder M.R, Corina D.L, Wright J.N. 1982. Mechanistic studies on C-19 demethylation in Estrogen Biosynthesis. Biochem. 201, 569-580.
15. Hackett J.C, Brueggemeier R.W, Hadad C.M. 2005. The final catalytic step of Cytochrome P450 Aromatase: A Density Functional Theory Study. J. Am Chem. Soc. 127, 5224-5237.
16. Van Landeghem A.A, Poortman J, Nabuurs M, Thijssen J.H. 1985. Endogenous concentration and subcellular distribution of Estrogens in normal and malignant Human Breast tissue. Cancer Res. 45, 2900-2906.
17. Brueggemeier R.W, Hachette J.C, Diaz-Cruz E.S. 2005. Aromatase Inhibitors in the Treatment of Breast Cancer. Endocr. Rev. 26, 331-345.
18. Brodie A. 2003. Aromatase Inhibitor Development and Hormone Therapy: a perspective, Semi Oncol. 30, 12-22.
19. Santen R.J Misbin R. 1981. Aminoglutethimide: A review of Pharmacology and Clinical Use. Pharmacotherapy, 1, 95-120.
20. Brueggemeier R.W. 2006. Update on the use of Aromatase Inhibitors in Breast Cancer. Exp Opin Pharmacotherapy, 7, 1919-1930.
21. Brodie A.M, Wing L.Y. 1987. In vitro and In vivo studies with Aromatase Inhibitor 4-Hydroxyandrostenedione. Steroids, 50, 89-103.
22. Santen R.J, Worgul T.J, Samojlik E, Interrante A, Boucher AE, Lipton A, Harvey HA, White DS, Smart E, Cox C , Wells SA. A randomized trial comparing Surgical Adrenalectomy with Aminoglutethimide plus Hydrocortisone in women with Advanced Breast Cancer. Engl. J. Med. 1981, 305: 545-551.
23. Plourde P.V, Dyroff M, Dowsett M, Demers L, Yates R, Webster A. Arimidex A.1995. New oral, once-a-day Aromatase Inhibitor. J. Steroid. Biochem. Mol. Bol. 53, 175-179.
24. Wang C, Makela T, Hase T, Adlercreutz H, Kurzer M.S. Lignans and flavonoids inhibit Aromatase Enzyme in human Preadipocytes. J Steroid Biochem Mol Biol 1994; 50: 205-212.
25. Narshimamurthy J, Rao RAR, Sastry GN. Aromatase inhibitors: A new paradigm in Breast Cancer, Curr. Med. Chem. Anti-Cancer Agents 2004; 4: 523-534.
26. Gobbi S, Cavalli A, Negri M, Schewe KE, Belluti F, Piazzi L, Hartmann RW, Recanatini M, Bisi A. 2007. Imidazolylmethyl Benzophenones as Highly Potent Aromatase Inhibitors. J. Med. Chem. 2007; 50, 3420-3422.
27. Bansal R. Narang G. Zimmer C. Hartmann R.W. 2011. Synthesis of some imidazolyl-substituted 2-benzylidene Indanone derivatives as potent aromatase inhibitors for breast cancer therapy, Med. Chem. Research, 20(6), 661-669.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT admin@pharmatutor.org
FIND OUT MORE ARTICLES AT OUR DATABASE








