
{ DOWNLOAD AS PDF }
ABOUT AUTHORS
MUNISH THAKUR*, Dr. ANUPAMA SETIA, Ms. NEETU.
DEPARTMENT OF PHARMACEUTICAL MANAGEMENT &
DRUG REGULATORY AFFAIRS,
JCDM COLLEGE OF PHARMACY,
SIRSA-HARYANA (INDIA).
munish.thakur98@gmail.com
ABSTRACT
The drug designated for production has to be manufactured in compliance with Current Good Manufacturing Practices (cGMP) following USFDA requirements, EU Directive or International Conference on Harmonization (ICH) Guidelines or Regulatory Authority of respective country. Regulatory authorities bear the responsibility to conduct inspections on pharmaceutical manufacturing plants to ensure they follows cGMP guidelines so that the drug manufactured is safe and effective. A quality system has to be set up such that the drug is manufactured in accordance with approved procedures. A drug is not permitted for sale until the marketing application for the new drug has been reviewed and approved by regulatory authorities. Extensive dossiers are provided to the authorities to demonstrate the safety, potency, efficacy and purity of the drug. After the drug has been approved and marketed, there is continuous monitoring of the safety and performance of the drug to ensure that it is prescribed correctly and adverse events (side effects) are investigated. The United States Food and Drugs Administration (FDA) has one of the most comprehensive and transparent regulatory systems in the world. In US Common Technical Document (CTD) format and most recently its electronic version-the electronic Common Technical Document (eCTD) format is used for submission of dossiers. Inclusion of a paragraph IV certification permits the Applicant to file its ANDA 4 years after the approval of a new chemical entity that is 1 year before the actual expiry of the 5 years exclusivity. In case patent exists that claims the drug, drug product, or method of use, the applicant is requested to file a patent certification with regards to the patent status. The different types of patent certifications are discussed. This project work elucidates US FDA’s previous interpretations of the statute regarding 180 days exclusivity and latest amendments in the current guidance. Information considered helpful in the compilation of different CTD modules 1, 2, 3, and 5 is discussed. Electronic submission in eCTD format is outlined.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-2534
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 5, Issue 11 Received On: 20/06/2017; Accepted On: 10/07/2017; Published On: 01/11/2017 How to cite this article: Thakur M, Setia A, Neetu; An Approval to manufacture and sale the copy of innovator product in the US Market; PharmaTutor; 2017; 5(11); 9-16 |
INTRODUCTION
Drug discovery occurs when a new compound is developed that addresses a specific disease target. After scientists identify a potentially viable drug candidate in the early drug discovery process, that compound proceeds to laboratory testing against specific targets to determine its effects. Most compounds are eliminated as they proceed from the discovery to the human development phase. When a chemical is found to impact a specific target, human testing is conducted through clinical trials to show the safety and efficacy of the chemical so that it may be approved by the FDA as a drug. The emphasis is that drugs should be safe, pure, effective and of consistent quality to ensure that they are fit to be used for their intended functions1. A drug is not permitted to sale until the marketing application for the new drug has been reviewed and approved by regulatory authorities. This process is performed within legislative framework which defines the requirements necessary for application to the concerned regulatory authority, details on the assessments procedure and the ground for approval or rejection of the application, and also the circumstances where a marketing authorization already granted may be withdrawn, suspended or revoked2. The authorities in The United States, The European Union and Japan asks for the common Technical Documents format and more recently, its electronic version the Electronic Common Technical Document.The current framework for generics in the United States is governed by the Drug Price Competition and Patent Term Restoration Act of 1984, more commonly known as Hatch-Waxman Act, and the Medicare Prescription Drug Improvement and Modernization Act (MMA) 3. The Hatch-Waxman created the section 505(j) of FD&C Act which was codified under title 21 of the CFR in section 314. Section 505(j) provides the statutory basis allowing pharmaceuticals companies to file Abbreviated New Drugs Application (ANDAs) instead of providing a full new drug application comprising the whole range of preclinical and clinical studies.
At the same time Hatch-Waxman allows brand name companies to apply for up to five additional years of patent protection for new medicines to compensate for time lost while their products were going through FDA’s approval process4.
Provisions of the Hatch-Waxman Act that protect products of innovator companies comprise:
A) Market Exclusivity upto 5 years
B) Patent Term Restoration
Benefits of the Hatch-Waxman Act for the generic manufacturer:
A) Implementation of Abbreviated New Drugs Application.
B) Establishment of Bioequivalence as the basis for approving generic drugs.
C) Providing manufacturer of generics with an exemption for pre expiry development and testing including the use of brand name drug to perform studies required for the approval process.
D) 180 days generic exclusivity.
With the publication of the Generic Drugs Final Rule and Initiative in June 20035, FDA revised its interpretation of the Hatch-Waxman Act by improving the implementation regulations with regards to generic drugs. The final rule facilitates the market entry of generic medicines by allowing a maximum of one 30-month stay per ANDA instead of previously possible multiple and overlapping 30 month stays. It clarifies the type of patents that must and must not be submitted to FDA for listing in Orange Book, revising the information required to be submitted on patents and consolidating all patent information on declaration forms to make those submissions are more informative and precise.
The Medicare Prescription Drug Improvement & Modernization Act which changed provisions of the FD&C Act that were originally added by the Hatch-Waxman Act.
Regarding Generics MMA provides for:
A) Single 30 month stay on a statutory basis.
B) First Commercial Marketing to trigger the 180 days exclusivity.
C) Forfeiture Mechanism to avoid scenarios where multiple first filers block one another to market.
D) Modification to be included in a single drug application on a statutory basis.
BASIS FOR SUBMISSION
The Legal basis for the filling of an ANDA is laid down in section 505(j) of the FD&C Act, which was implemented by Hatch-Waxman Act. Regulatory action required for the submission of an ANDA by the applicant is provided in the corresponding section of CFR, that is subpart c or section 314.62 to 314.99. The content and format of and abbreviated applications described in section 314.94 of the CFR. An ANDA is usually submitted for a drug product that is the same as a drug product previously approved (Reference Listed Drug) by the FDA.
Application submission is generally substantiated by the following particulars
A) Reference to the name of the Listed Drug,including gits dosage for m and strength
B) Astatementasto whether the Listed Drug is entitled to aperio do f marketing exclusivity
C) However,asstipulatedin§314.92anAbbreviatedNewDrugApplicationmay only be submitted for drug products that are thesameasthe(Reference)Listed Drug.As further detailed in § 314.92(a)(1) theterm"same as"means, the drug product and Listed Drug6:
(i) Shall contain the identical activeingredients
(ii) Shall beidentical in strength, dosageform, androute of administration
(iii) Shall haveidentical conditions of use
MARKET EXCLUSIVITY
New drug exclusivity is implemented by provision sunder section 505(j)(5)(F) of the Act codifiedin21CFR314.108. This section of the Act provides for specific time periods, known as new drug product exclusivity or market exclusivity, during which an ANDA can not be submitted or the effective date of approval for an ANDA must be delayed.Aspersection505(c)(3)(E) and 505(j) (5) (F) of the Act a 5-year period of exclusivity is granted for new chemical entities (NCE) not previously approved by FDA.
The selections of the Act expressly state that no ANDA may be submitted during the exclusivity period an d that such applications may be submitted after 4 years only if they contain a certification of patent in validity or non- infringement. Under the same sections it islaid down that certain drugs or changes to drugs such as a change in dosage form or strength, new indications or routes of administrationcanreceive 3 years periodof exclusivity. However, approval of three-year exclusivity requires new clinical investigations (other than bioavailability studies).
PATENT PROTECTION The patent confers"theright to exclude others from making, using, offering for sale,or selling the in vention through out the United States or importing the in vention in to the United States” Patent applications are filed with the USPTO, which is part of the US Department of Commerce. Hence, the role of the USPTO is only to grant patents or to register trademarks but not to protect theinventions. Generally, the period of validity of a new patentis 20 years from the date offiling of the patent application7. Patents may only been forced vialawsu its. If a patent is in fringed, the patentee may sue forrelief in the appropriate federalcourt. The patentee may ask the court for a restriction to prevent the continuation of in fringement and may also ask the court for an award of damages because of the in fringement. In such an in fr in gement suit, the defend an tmayra is e the question of the validity of the patent, which is then decided by the court. Patent Term Restoration (PTR) enacted by the Hatch-Waxman Act in1984 compen sates patent holders for marketing time lost while developing theproduct and awaiting FDA approval by extending the patent term up to five years.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
According to section 505(j) (2) (vii) of the Act ANDA sare required to include certifications on the status of all patents applicable to the listed drug. Section §314.94(a)(12) of theCFR describes the patent certification requirements for ANDAs ainterpreted by the FDA. Section314.94 presents an overview on the provisions that will be discussed in turn:§ 314.94(a) (12)6.
A) Patents claiming drug, drugproduct, or methodof use
B) No relevant patents
C) Method of usepatent
D) Method of manufacturing patent
E) Licensing agreements.
Types of Patent Certification and Effects on ANDA Approval
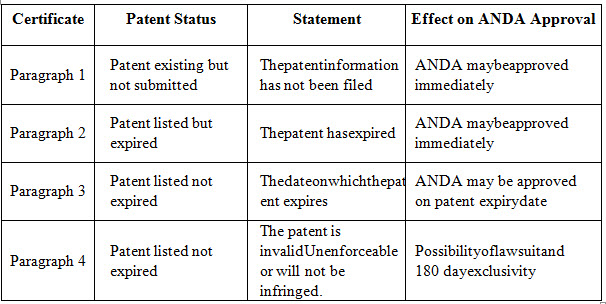
30-MONTH STAY
The ANDA applicant who has in cludeda Paragraph IV Certification in its application is required to send a 'Noticeof certification of invalidityor non-infringement of apatent' also known as 'notice of certification' to each owner of the patent in question and to the holder of the approved NDA to which the ANDA refers.
Section314.95 of the CFR descry best he current require ments with regard to the sending of the notice and the information to be included in the notice by the applicant. According to tha tregulation the applicant tisobliged to sendthe notice notonsubmission of the ANDA but when it receives from FDA an acknowledgement letter stating that its ANDA is sufficiently complete to permit are view of the entire dossier. If the innovator company decides to sue the genericapplicant for the patent infringement, it must do so within 45 daysfrom receipt of notice according to section505(j)(5) (B) (iii)of the Act. Insuchcases, the FDA will delay the approval of the ANDA upto 30 months for pending resolutionoflaw suit.FDA approvalcan only come into effect when the 30-months period haselapsed, orthegeneric applicant wins during patentlitigation. As outlines in the following the MMA introduced significant improvement in the field of ANDAs, e.g. by providing a statutory basis for a single 30-month stay and benefits in terms of timing of ANDA approvals.
180 DAY EXCLUSIVITY
Section 505(j)(5)(B)(iv) of the Act provides an incentive for generic manufacturers filing a Paragraph-IV Certification to challenge patents that may be invalid, not infringed orunen force able, thereby possibly triggering a patent action against them by the patent owner. According to this section of the Act the first generic applicant to submit an application containing a Paragraph IV Certification will enjoy 180 days of generic exclusivity, since all subsequent applications, although including a Paragraph IV Certification will be made effective on the date that is 180 days after the date of the first commercial marketing of the drug by the first applicant. In situations where different applicants challenged different Orange Book-listed patents for the same drug product FDA adopted a complicate" shared exclusivity “approach hunder which the first-to-file applicants with a ParagraphIV Certification for eachlisted patent shared the180 day exclusivity period based on that patent. Once the first-to-file applicant for feits its180-day exclusivity, no subsequent ANDA applicant will be eligible for180- day exclusivity. Any subsequent ANDA filer may launch its generic product immediately provided that any patent is sues are resolved and final FDA approva lis received8
SUPPLEMENTS In case a change to the product applied for would lead to a significant difference as compared to the listed drug cited in the initial submission (e.g., different active ingredient, dosage form, route of administration) a new application should be filed for the different drug product and the new ANDA should refer to the separate listed drug with the desired characteristics (e.g., active ingredient, dosage form, route of administration) if identifiable in the Orange Book. The applicant is not allowed to submit a supplement or amendment to its pending or approved application to seek approval for such a change. Generally, a single application can be used to seek approval for different strengths of the same listed drug. Consequently, an applicant may submit an amendment or supplement to seek approval of a different strength from that for which the application was initially submitted and is not required to file a separate application for such a change. This is expressly permitted under the Act, as amended by the MMA (see section 505(j) (2) (D) (ii) of the Act).
CONTENT ANDFORMAT OF ANDA6
|
ArchivalCopy-Requirement/ Title acc. to21CFR§ 314.94(a) |
ReviewCopy |
FieldCopy |
Location within CTD*** |
|
|---|---|---|---|---|
|
Chem.* |
Bio. ** |
|||
|
(1)Application form. |
X |
X |
X |
Module1 |
|
(2)Table of contents. |
X |
|
|
Module1 |
|
(3)BasisforAbbreviatedNew Drug Application submission |
X |
X |
|
Module 1 |
|
(4)Conditions of use |
X |
|
|
Module1 |
|
(5)Activeingredients. |
X |
|
|
Module1 |
|
(6)Route of administration,Dosageform,and strength. |
X |
|
|
Module 1&5 |
|
(7)Bioequivalence. |
|
X |
|
Module 2&5 |
|
(8)Labelling. |
X |
X |
|
Module1 |
|
(9)Chemistry, manufacturing, and controls. |
X |
|
X |
Module 2&3 |
|
(10)Samplestatement required under §314.50(e) (1)and methods validation package as per (e) (2)(i). |
|
|
|
Sample on request |
|
(11)Otherinformation as described in §314.50(g),e.g. referenceto information submitted previously. |
|
|
|
Module2 &3 as applicable |
|
(12)Patent certification |
X |
|
|
Module1 |
Chemistry/microbiology **, Bioequivalence, ***Common Technical Document When paper format is submitted, the applicant is requested to submit three copies of the application accordingto§314.94(d).
Module 4- Non Clinical Part:Module 4 can be omitted for ANDAs.
Module 5-Clinical Part:
The format of the clinical part follows the ICH-CTD standard for Module 5 as described in the M4E (R1) Guideline. ICH efficacy guidelines regulate general aspects on clinical trials such as
E3 Structure and Content of Clinical Study Reports
E6 (R1) Good Clinical Practice
E8 General Consideration of Clinical Trials
E9 Statistical Principles for Clinical Trials
E10 Choice of Control Group and Related Issues in Clinical Trials
E11 Clinical Investigation of Medicinal Products in the Pediatric Population
E12 Guidelines for Clinical Evaluation by Therapeutic Category
Specific bioavailability (BA) / bioequivalence (BE) requirements are subject to national regulations in the US. According to section 314.94(a)(7)(i) the ANDA applicant is obliged to submit information showing that the proposed drug product is bioequivalent to the Reference Listed Drug to which reference is made. Regulatory requirements for documentation of bioequivalence are provided in part 320 of the CFR.
SubmissioninElectronic Format-eCTD:
The eCTD format is an electronic standard that provides a harmonized technical solution for the transfer of regulatory information contained in the Common Technical Document from Industry to Agencies across ICH region. The information to be included in the technical sections of Modules 1 to 5 follows the provisions laid down in the ICH M4 Guidelines. The focus of the specification is to provide the ability to transfer the registration application electronically from industry to a regulatory authority while at the same time also facilitating the creation, review, lifecycle management and archival of the electronic submission. The specification developed by the ICH M2 EWG was the Electronic Common Technical Document Specification, which is based on Extensible Mark-up Language (XML) technology and lists the criteria that will make a electronic submission technically valid. Based on ICH CTD and eCTD specifications the FDA has published Guidance on "Providing Regulatory Submissions in Electronic Format -Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications" which provides recommendations on how to organize electronic applications to FDA10. This guidance refers to a series of technical specifications which should be followed for the preparation of an electronic application in accordance with the eCTD format. Applicants may make a sample submission reviewed by FDA to resolve any technical issues with the eCTD submission prior to the actual submission. The sample submission is however not considered an official eCTD submission. Before submission of the actual ANDA the applicant should request a "Pre-Assigned Application number". A step-by-step description of the procedure to be followed is provided on the web site of the FDA.Applicants who would like to submit electronically, but who are unable to submit in eCTD format by the official start-date, can ask for an eCTD waiver.
A) Archival Copy: The archival copy is a complete copy of an application and is intended to serve as the official referene resource for the Agency.Thearchival copy is maintained during there view of the application to permit in divid ualre viewers to refer to in formation that is not contained in their particular technical sections of the application.
B) Review Copy: There view copy is used to evaluate the application.Itisusually divided into two parts containing the scientific information needed for chemistry/microbiology review an dbioequivalencere viewc on ducted by different scientificre viewers.
C) Field Copy: The field copy is used to enable pre-approval inspection by the FDA. The field copy should hence contain the technical section of the chemistry, manufacturing and controls part for drug substance and drug product according to section 314.50(d)(1) as required in section 314.94(a)(9) of the CFR.
FDA encourages applicants to submit their ANDAs in the CTD format and preferably as electronic CTD. The Common Technical Document is divided into four separate sections. The four sections address the application organization (M4), the Quality section (M4Q), the Safety section (M4S) and the Efficacy section (M4E) of an application9.
Module 1-Regional Requirements:
The content and format to be followed in the preparation of individual documents of Module 1can be obtained from current FDA presentations published on the homepage of FDA or the Generic Pharmaceutical Association(GPhA).
Module 2-Overview and Summaries:
Module 2 contains the Summary of Module 3 as well as the Overview and Summary of Module 5. Non-clinical, toxicological information and non-clinical study reports located in Module 4 can be omitted for ANDAs.
Module 3-Chemistry, Manufacturing and Control:
The content and format of the chemistry, manufacturing and control spartmainly issu bject to ICH Guidelines. The format follows the ICH-CTD standard for Module 3asdescribedinthe M4Q(R1) Guideline. The majority of ICH Quality guideline shave mean while been adopted by FDA. Despite this common bas is a few regional spects specific to FDA are to be considered for the compilation of Module3.
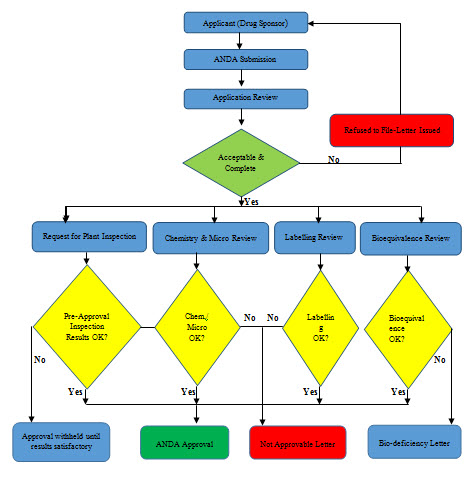
PROCESS OF ANDA APPROVAL:
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESULTS
The CTD is organized into five modules. Module 1 is designed to contain region-specific information such as application forms and other administrative provisions that may apply. As such, it is not harmonized and is not considered part of the CTD. The other four modules present the technical data in a harmonized format. Module 2 should contain critical overview assessments of the quality, non-clinical and clinical data, together with summaries of the non-clinical and clinical data. The objective of this section is to provide reviewers with an introduction to the submission, and to orient them as to how the applicant believes the data support the granting of a marketing authorization. Modules 3 provide detailed quality data. Module 4 which, is for Nonclinical Study Report is not required for ANDAs. Module5 provide the detailed clinical data.
Hatch-Waxman provides that an applicant for an ANDA or a 505(b)(2) applicant must make an appropriate certification respecting marketing of the generic drug. In an effort to encourage generic drug entry into the pharmaceutical market, the Hatch-Waxman Act provides a commercial incentive to those generic companies who are the first to file ANDAs incorporating Paragraph IV Certifications challenging infringement or validity of Orange Book–listed brand-name pharmaceutical patents. Thus, in the event the listed brand-name patent(s) are adjudged to be invalid or not infringed prior to their normal expiration date(s), the first generic filer entitled to approval of its ANDA receives a 180-day exclusivity period, during which no other ANDA can be approved for the same product. In such circumstances, the first entity to file an ANDA can be the only generic marketer for 180 days.
CONCLUSION:
In the United State, applications to market a generic are filed by so called “Abbreviated New Drug Applications (ANDA)”. The application is called abbreviated because results from (pre) clinical safety and efficacy studies are usually not required. Instead, reference is made to the data on file for an already approved reference drug. The legal basis for the filing of an ANDA is laid down in section 505(j)of the Food, Drugs & Cosmetics Act which is codified in 21 CFR 314. An ANDA usually is submitted for a drug product that is the same as a drug product previously approved by the FDA. The approved drug product, usually an innovator drug, when referred to in a generic application, is called the Reference Listed Drug. In case a patent exists that claims the drug, drug product, or method of use the applicant is requested to file a patent certification with regard to the patent status. The different types of patent certifications are available. Inclusion of a Paragraph IV Certification permits the Applicant to file its ANDA 4 years after approval of a new chemical entity that is 1 year before actual expiry of the 5 year exclusivity. The submission of an ANDA containing a Paragraph IV Certification is an infringing act and, therefore, may be followed by a patent infringement litigation. In such cases, the FDA will delay the approval of the ANDA up to 30 months for pending resolution of lawsuit. As provided by the Food, Drugs & Cosmetics Act, the first generic applicant to submit an application containing a Paragraph IV Certification is eligible to an incentive of 180 days of generic exclusivity. This exclusivity, commonly known as "180-day exclusivity", protects the first- to-file applicant, whose ANDA contains a Paragraph IV Certification, from competition by subsequent generic applicants referring to the same Reference Listed Drug.
REFERENCES
1.RickNg. Drugs From Discovery To Approval, A John Wiley&Sons Inc.Publication, Hoboken, New Jersey,2004.
2. http://en.wikipedia.org/wiki/Marketing_authorization
3. www.regulatoryone.com/2012/01/anda.html
4. CDER,Office of Generic Drugs web site,https://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/ucm119100.htm
5. Generic Drugs Final Rule and Initiative, June 2003
6. 21 CFR 314 at the U.S.Government Printing Office web site https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=314
7.Frequently Asked Questions on the Patent Term Restoration Programon FDA website,
https://www.fda.gov/drugs/developmentapprovalprocess/smallbusinessassistance/ucm069959.htm
8. 180-Day Generic Drug Exclusivity on FDA website, https://www.fda.gov/drugs/developmentapprovalprocess/smallbusinessassistance/ucm069964.htm
9. CTD on ICH website http://www.ich.org/products/ctd.html
10.ICH- eCTD Specification and Related Files,http://estri.ich.org/eCTD/index.htm
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








