
About Author
Mohd Riyaz Beg1* 1
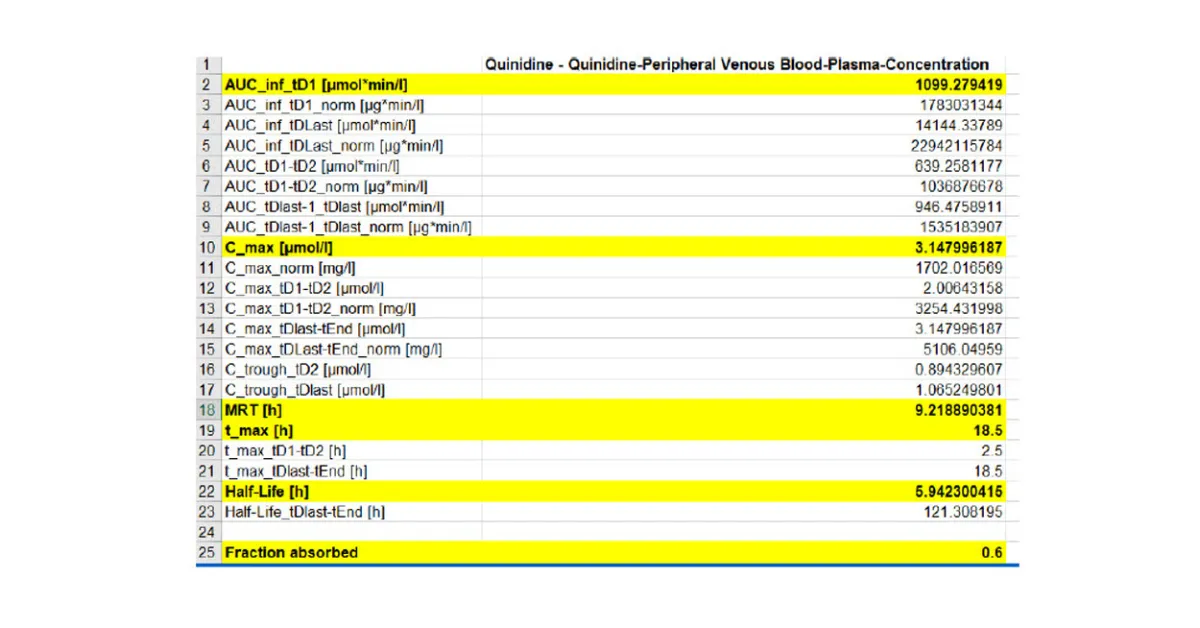
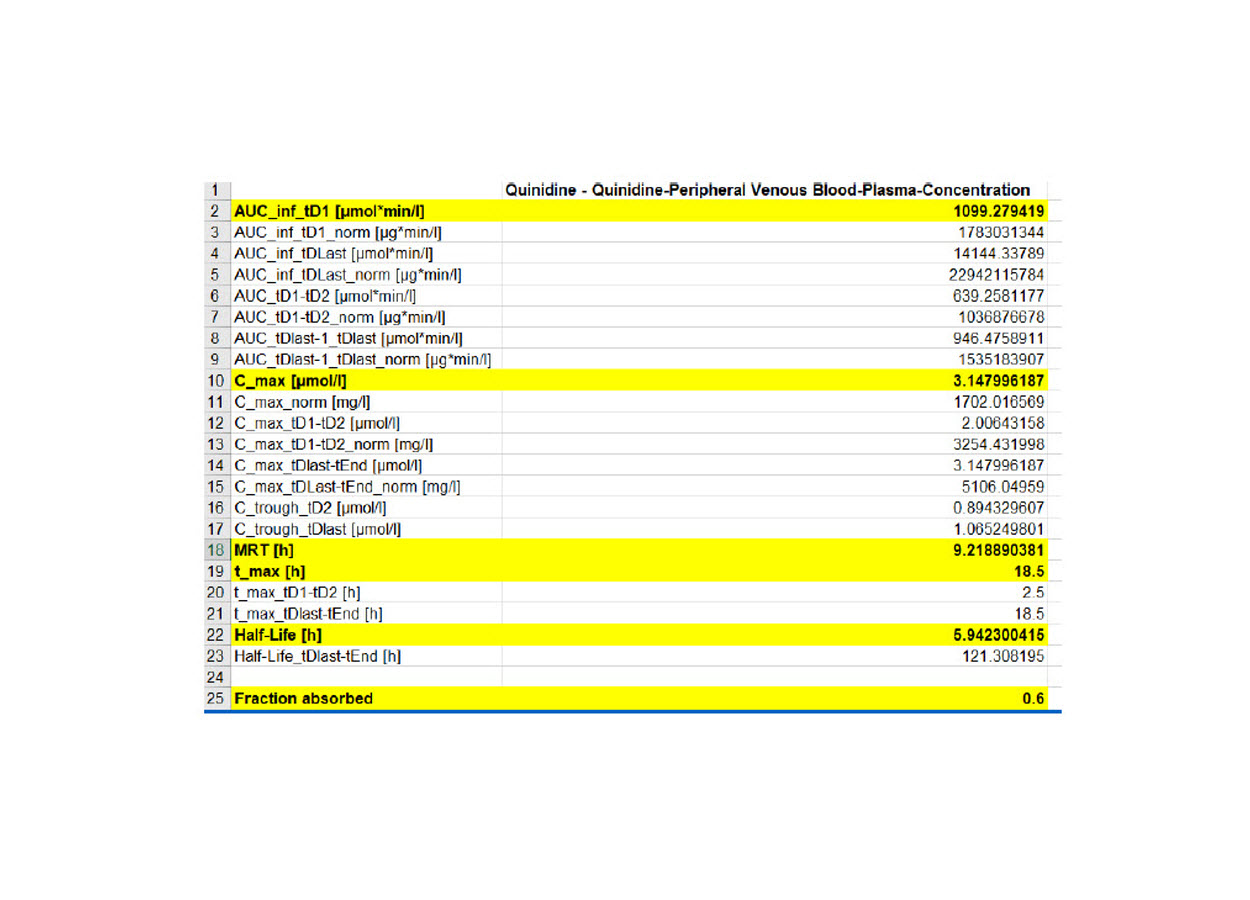
Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Mumbai
*Corresponding author’s
email: phm19mr.beg@pg.ictmumbai.edu.in; mohdriyazbeg@gmail.com
ABSTRACT
This article takes you through the possible pharmacokinetic models and parameters for the antiarrhythmic drug Quinidine. The shuffle between the linear and non-linear pharmacokinetics of quinidine make it a candidate for therapeutic drug monitoring. The adverse effect in the form of QT-elongation is one of the major challenges to go for estimating good kinetics of the quinidine. The current article helps in providing a wide perspective towards the available models for the quinidine's ADME and compile the information published by several authors to help you to understand the quinidine pharmacokinetic behaviour.
Commentary
Quinidine, a very old drug exploring since 1749 for its antiarrhythmic activity. When we talk about the activity of any drug in the body, the first thing that we need to look forward is the ADME to make that drug eligible to reach the site of the action from the site of administration. This ADME (aka Pharmacokinetics) deals with a mathematical description of the rate of drug movement into, within, and exit from the body. It also includes the biotransformation and drug metabolism. Quinidine is a BCS class I agent with high solubility and permeability hence, show an oral bioavailability of 70%. The apparent volume of distribution, V is 160 L to 280 L. Once it makes its way in the plasma it shows a significant amount of serum lipoproteins binding. It is 70% to 90% bound to plasma protein, primarily to albumin but also to many other plasma constituents such as α-acid glycoprotein (because chemically it is diprotic acid). It has an elimination rate constant, Ke, 0.06 hr-1 to 0.14 hr-1. The clearance, Cl, ranges from 12 L/hr to 24 L/hr. Oral quinidine is available either as relatively rapidly adsorbed conventional tablets (usually quinidine sulphate) or as a variety of slowly adsorbed sustained-release preparations. The fraction available, F, is generally 0.7 or greater. Values of the first-order absorption rate constant, k, range from 0.63 hr-1 ~ to 2.97 hr-1. Evidence of dependence of F or Ka on dosage form is conflicting. The main conflicting scenario with Quinidine is that have a variety of adverse effects in which QT-interval prolongation is the major one which is dose-dependent. Similarly, it requires dose modulation in geriatric patients and patients suffering from cirrhosis, hypoalbuminemia, and renal insufficiency. To make it relatively safe to use and improve its therapeutic modalities we need to understand its kinetics in the body.
As we know that PK of Quinidine gets influenced by severe cardiac, liver failure and renal function impairment. We had tried to look upon several models to get the best fit for its kinetics. Quinidine’s PK model mapping is not an easy task to perform. Different studies showed different results with variety of factor related to PK of Quinidine. Some researchers proposing that Quinidine exhibits linear pharmacokinetics whiles others have suggested that its disposition is nonlinear. A famous study by Bolme & Otto, 1977, showed a non-linear relationship between quinidine dose and steady-state plasma drug concentration. Furthermore, the steady-state drug concentration which is devised from single-dose data is found to be consistently higher. In addition to this, another study suggested that Quinidine kinetics can be best described by a two-compartment model with zero-order absorption from the gastrointestinal tract. To answer the consistently high dose to attain steady state, we can say that increasing the dose of quinidine may result in a disproportionate increase in steady-state plasma conc. in some individuals. Through the keen observation of the plasma conc. vs time profiling we can say with a certain degree of confidence that its disposition is non-linear and it demonstrates dose-dependent kinetics when administered orally. The two Quinidine metabolites found in plasma, 3-hydroxyquinidine and 2’-oxoquinidinone, have been also measured to determine whether changes in the metabolism could account for the apparent non-linear PK with increasing dose. It is also noted that in many cases where no satisfactory fit with the one and two-compartment models could be achieved, the plasma profile did not follow linear mammillary model kinetics. After the distribution phase, with both intravenous and oral administration, a shoulder (Bulge) is frequently observed in the plasma concentration curve. The same trend can also be found in curves published by Ueda et al., although these authors do not comment on it. Possible explanations for it are enterohepatic cycling or nonlinearity in quinidine disposition as it more towards the biphasic type of curve fitting. With our limited understanding of its multiple-dose kinetics, we may require to explore a three-compartment model. Where a two-compartment solution does not appear to come noticeably closer to a satisfactory fit than a one-compartment solution. A three-compartment model, however, provided a marked improvement in fit in the plasma conc. vs time profile. To ensure goodness of fit in the curve, we need to consider inter- and intrasubject variation in the drug disposition with least square fitting procedures. Simultaneously, the two-compartment model with zero-order input is preferred over one- and two-compartment models with first-order input and a slightly improved fit resulted by the introduction of lag time as a parameter. In case of elimination, a biexponential decline in the log plasma quinidine concentration vs time curves is observed. Very interestingly, dose-dependent elimination of quinidine and that the variability in quinidine’s pharmacokinetics is related in part to its hydroxylation (the metabolism step). Deeming to the population PK model that best described the PK of Quinidine in plasma was a 2-compartment model with linear absorption and elimination and lag time for absorption.
With increasing interest in IVIVE, micro-dosing and PBPK studies, that enabled us to gain a plausible mechanistic explanation for the observed ethnic differences in QTc prolongation, despite the similarities in the measured plasma concentrations in different individuals. Micro-dosing studies allow us to obtain direct pharmacokinetic data for compounds in humans before clinical trials, without having to perform laborious nonclinical toxicity studies. One of the major concerns with micro-dosing studies is the nonlinearity of the pharmacokinetics between micro-dose and therapeutic dose caused by the saturation of metabolic enzymes and transporters. Quinidine is the model drugs showing nonlinear pharmacokinetics because it is a good substrate of MDR1 as well as of CYP3A4. So, the possible mechanism underlying this nonlinearity could be the saturation of the MDR1-mediated efflux and/or CYP3A4- mediated metabolism in the small intestine.
PK study using PKSim
To access the full Plasma conc. Vs Time data :
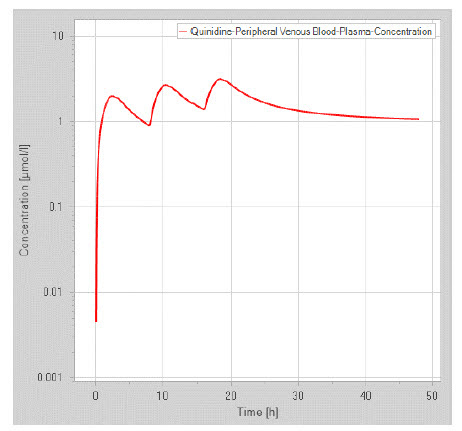
Figure 1: Plasma conc. vs time profile predicted by PKSim
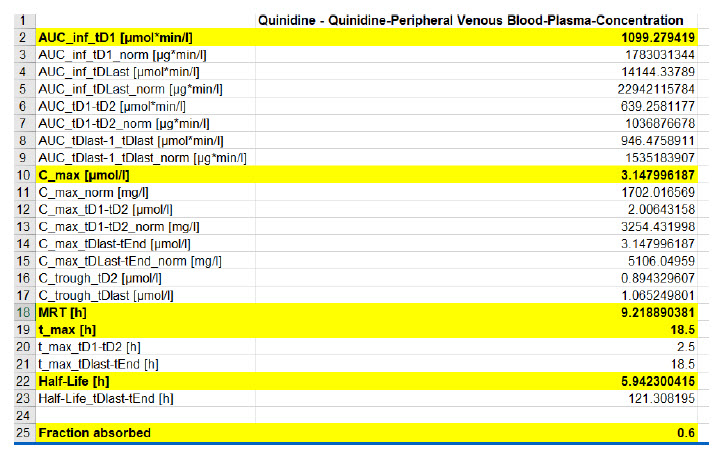
Figure 2 : Predicted PK data using PKSim
Conclusions
To the end this discussion, we can say that further evaluation is necessary with powerful statistical techniques like Smooth Nonparametric methods and computational mathematical models consisting the QSAR module to prove a better understanding of PK parameters of Quinidine. As per the improvement in PD-models like IC50 (based on Hills eqn.) and Emax we can predict a better kinetic mapping of the ADME of Quinidine to market this wonderful drug with highest safety and efficacy.
References
1. Brosen, K., Davidsen, F., & Gram, L. F. (1990). Quinidine kinetics after a single oral dose in relation to the sparteine oxidation polymorphism in man. British journal of clinical pharmacology, 29(2), 248-253.
2. Chetty, M., Rose, R. H., Abduljalil, K., Patel, N., Lu, G., Cain, T., ... & Rostami-Hodjegan, A. (2014). Applications of linking PBPK and PD models to predict the impact of genotypic variability, formulation differences, differences in target binding capacity and target site drug concentrations on drug responses and variability. Frontiers in pharmacology, 5, 258.
3. Crevasse, L. (1988). Quinidine: An update on therapeutics, pharmacokinetics and serum concentration monitoring. The American Journal of Cardiology, 62(14), I22-I23.
4. Davidian, M., & Gallant, A. R. (1992). Smooth nonparametric maximum likelihood estimation for population pharmacokinetics, with application to quinidine. Journal of Pharmacokinetics and Biopharmaceutics, 20(5), 529-556.
5. Fattinger, K., Vozeh, S., Ha, H. R., Borner, M., & Follath, F. (1991). Population pharmacokinetics of quinidine. British journal of clinical pharmacology, 31(3), 279-286.
6. Grenier, J. (2010). Population Pharmacokinetic Meta Analysis: Inhibition by Quinidine of the First-Pass and Systemic Metabolism of Dextromethorphan to Dextrorphan J. Grenier (1), JR. Lavigne (1), LE. Pope (2)(1) Clinical Pharmacology Department, Celerion, Montréal, Qc, Canada;(2) Avanir Pharmaceuticals, Aliso Viejo, CA, USA.
7. Guentert, T. W., Holford, N. H., Coafes, P. E., Upton, R. A., & Riegelman, S. (1979). Quinidine pharmacokinetics in man: choice of a disposition model and absolute bioavailability studies. Journal of pharmacokinetics and biopharmaceutics, 7(4), 315-330. 8. Maeda, K., Takano, J., Ikeda, Y., Fujita, T., Oyama, Y., Nozawa, K., ... & Sugiyama, Y. (2011). Nonlinear pharmacokinetics of oral quinidine and verapamil in healthy subjects: a clinical microdosing study. Clinical Pharmacology & Therapeutics, 90(2), 263-270.
9. Ochs, H. R., Greenblatt, D. J., & Woo, E. (1980). Clinical pharmacokinetics of quinidine. Clinical pharmacokinetics, 5(2), 150-168.
10. Ochs, H. R., Greenblatt, D. J., Woo, E., Franke, K., Pfeifer, H. J., & Smith, T. W. (1978). Single and multiple dose pharmacokinetics of oral quinidine sulfate and gluconate. The American journal of cardiology, 41(4), 770-777.
11. Polak, S. (2013). In vitro to human in vivo translation–pharmacokinetics and pharmacodynamics of quinidine. ALTEX-Alternatives to animal experimentation, 30(3), 309-318.
12. RUSSO Jr, J. O. H. N., RUSSO, M. E., SMITH, R. A., & PERSHING, L. K. (1982). Assessment of quinidine gluconate for nonlinear kinetics following chronic dosing. The Journal of Clinical Pharmacology, 22(5?6), 264-270. 13. Wooding?Scott, R. A., Smalley, J., Visco, J., & Slaughter, R. L. (1988). The pharmacokinetics and pharmacodynamics of quinidine and 3?hydroxyquinidine. British journal of clinical pharmacology, 26(4), 415-421.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT admin@pharmatutor.org
FIND OUT MORE ARTICLES AT OUR DATABASE








