

 About Authors:
About Authors:
V.DEEPTHI *, POORNIMA.Y, DR.G.DEVALA RAO, T.SANDEEP REDDY
Department of Pharmaceutical Analysis,
K.V.S.R.Siddharthacollege of pharmaceutical sciences,
Vijayawada-520010, India.
*deepthi759@gmail.com
ABSTRACT
A novel stability-indicating RP-HPLC method has been develop and validated for quantitative analysis of Sitagliptin in the bulk drug and in its pharmaceutical dosage forms using Hypersil–BDS- C18 column (250x4.6mmi.d, 5µ particle size) with 10mM Phosphate buffer (PH-3.5): ACN 60:40%v/v as isocratic mobile phase enabled separation of the drug from its degradation products. UV detection was performed at 260 nm. The method was validated for linearity, accuracy (recovery), precision, sensitivity, ruggedness and robustness. The linearity of the method was excellent over the range 10–60μg/ml (correlation coefficient 0.999). The limits of detection and quantification were 0.21 and 0.640μg/ml, respectively. Recovery of Sitagliptinfrom the pharmaceutical dosage form ranged from 99.99 to 100.05%.
Sitagliptin was subjected to stress conditions (Hydrolysis (acid, base), oxidation,thermal and photo degradation) and the stressed samples were analysed by use of the method. Degradation was observed in acid, base, and 30% H2O2. The drug was stable under the other stress conditions investigated. The degradation products were well resolved from main peak. The forced degradation studies prove the stability indicating power of the method.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1529
INTRODUCTION
Sitagliptin1,2 is a new oral hypoglycemic (anti-diabetic drug) of the new dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs. This enzyme-inhibiting drug is to be used either alone or in combination with metformin or a thiazolidinedione for control of type 2 diabetes mellitus. The drug works to competitively inhibit a protein/enzyme, dipeptidyl peptidase 4 (DPP-4), that results in an increased amount of active incretins (GLP-1 and GIP), reduced amount of release of glucagon (diminishes its release) and increased release of insulin.
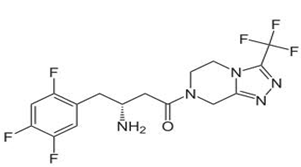
[adsense:468x15:2204050025]
Fig. 1: structural formula of Sitagliptin
Stability testing forms an important part of the process of drug product development. The purpose of stability testing is to provide evidence on how the quality of a drug substance or drug product varies with time under a variety of environmental conditions, for example temperature, humidity, and light, which enables storage conditions, retest periods, and shelf life to be recommended.The two main aspects of study of the stability of a drug product that play an important role in shelf life determinations are assay of the active drug and the degradation products generated during stability studies. Assay of a drug product in a stability test sample must be performed with stability?indicating method, as recommended by the International Conference on Harmonization (ICH).
A literature survey revealed that very few methods3,4,5,6,7,8 were developed and none of the reported procedures enables analysis of the Sitagliptin in pharmaceutical dosage forms in the presence of their degradation products. This manuscript describes the development and validation, in accordance with ICH guidelines,ofarapid,economical, precise, and accurate stability?indicatingisocratic RP?HPLC method for analysis of Sitagliptininthepresence of its degradation products.
MATERIALS AND METHODS
Chemicals and solutions
An analytically pure sample of Sitagliptin (purity 99.7%) wasprocured as gift sample from Granules pharma (Hyderabad, India) and Tablet formulation [JANUVIA (Brand name), Granules pharma, Hyderabad, India] was procured from a localpharmacy with labelled amount 50mg. Acetonitrile and Methanol (HPLC grade) were obtained from Merck Fine Chemicals(Mumbai, India), Potassium Di-hydrogen phosphate (AR grade) from Loba chemicals ( Mumbai, India), sodium-hydroxide (NaOH), hydrochloric acid (HCl), and hydrogen peroxide(H202) were from Qualigens Fine Chemicals (Glaxo, Mumbai, India).The 0.45?μm Nylon pump filter was obtained from AdvancedMicrodevices (AmbalaCantt., India). Doubledistilled water was usedthroughout the experiment. Other chemicals used were of analyticalor HPLC grade.
Preparation of standard solutions
Accurately weigh and transfer 10 mg of SitagliptinWorkingstandard into a 100 ml volumetric flask, add about 50 ml of Methanol (Diluent)andsonicate to dissolve it completely and make volume up to themark with the same solvent (100μg/ml, Stock solution). Furtherpipette 1 ml of the above stock solution into a 10ml volumetric flaskand dilute up to the mark with Mobile phase (diluent), it was 10μg/ml. Standardcalibration solutions (10–60μg/ ml) for assessment of linearitywere prepared from this stock solution by dilution with suitablediluent.
Preparation of sample solutions
The commercially available Tablet contains Sitagliptin (100mg). 20 tablets were weighed individually and finely powdered. A powder blend equivalent to 10mg of SIT was transferred to a 10mL volumetric flask containing 10 ml of theMethanol(diluent) and sonicated for 5min. and then final solution was made with diluent to get thesolution of 100μg/ml From this solution, 1 ml was taken in 100ml standard volumetric flask and diluted to 100 ml withMobile phase(diluent)togive a solution of 10μg/ml From this stock solution, variousdilutions of solution were prepared and analysed.
Chromatography
The liquid chromatographic system consisted of followingcomponents: Shimadzu HPLC model containing LC?20AT (VP series)pump, variable wavelength programmable UV/ VIS detector SPD?20A (VP series) and Hamilton syringe (705NR,50μl).Chromatographic analysis was performed using Empower -2 software on a Hypersil–BDS-C18 column with 250 x 4.6 mmi.d, 5μm particle size.The optimized mobile phase was consisted of 10mM Phosphate buffer (PH-3.5): ACN with 60:40%v/v .The flow rate was 1.2ml / min. Detection wavelength 260 nm was selected by scanning drug over a wide range of wavelength 200 nm to 400 nm in spectrophotometer. The 20μl sample was injected and the total run time was 6 min. Chromatogram showed peak of Sitagliptinat retention time of 2.157 ± 0.02 min.
Forced degradation study
To study the effect of acid, approximately 100 mg Sitagliptinwas accurately weighed and dissolved in 25 ml of 1M hydrochloric acid (HCl) and refluxed for 70°C for approximately 2 hr in water bath. The solution was then left to reach ambient temperature, and neutralized to pH 3.5 by addition of 1M sodium hydroxide( NaOH) then diluted to 50 ml with diluent (Mobile Phase) from this solution target concentration was prepared and injected.
To study the effect of alkali, approximately 100 mg Sitagliptin was accurately weighed and dissolved in 25 ml of 1M sodium hydroxide(NaOH) and refluxed for 70°C for approximately 2 hr in water bath. The solution was then left to reach ambient temperature, and neutralized to pH 7 by addition of 1M hydrochloricacid (HCl) then diluted to 50 ml with diluent (Mobile Phase) from this solution target concentration was prepared and injected.
To study the effect of oxidising conditions, approximately 50 mgSitagliptin was accurately weighed and dissolved in 25 ml of30% H2O2 and refluxed for 40°C for approximately 2 hr in waterbath. The solution was then left to reach ambient temperature, anddiluted to 50 ml with diluent (Mobile Phase) from this solutiontarget concentration was prepared and injected.
To study the effect of photolytic effect, approximately 50mgSitagliptin was accurately weighed and exposed to UV light for 24 hours. Then the drug substance was made to 100ug/ml with diluent (mobile phase) and refluxed for 70°C for approximately 2 hrinwater bath. The solution was then left to reach ambient temperature, from this solutiontarget concentration was prepared and injected.
To study the effect of temperature, approximately 50 mgSitagliptin was stored at 80°C for 48 hr, then dissolved in fewml of diluent and sonicate for 5min then diluted to 50 ml withdiluent (Mobile Phase) from this solution target concentration wasprepared and injected.
Method validation
The method was validated according to International Conference on Harmonization 9,10guidelines for validation of analyticalprocedures.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESULTS AND DISCUSSION
System suitability
System?suitability tests are an integral part of method developmentandare used to ensure adequate performance of thechromatographic system. Retention time (Rt), number of theoreticalplates (N), tailing factor (T), and peak asymmetry (Af) wereevaluated for five replicate injections of the drug at a concentrationof 30μg / ml. The results given in Table 1 were with-in acceptablelimits. A typical chromatogram of Sitagliptin is presented infig 2.
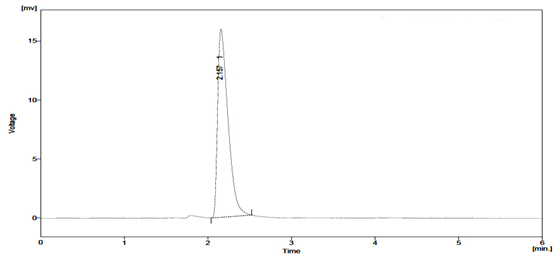
Fig. 2: Chromatogram of Sitagliptin formulation at 260 nm
Table 1: Results from system suitability studies
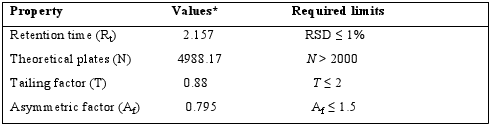
* Mean ± S.D. from six determinations
Table 2 : Calibration data of Sitagliptin by RPHPLC method
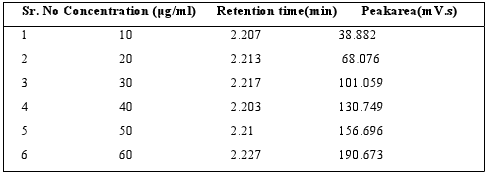
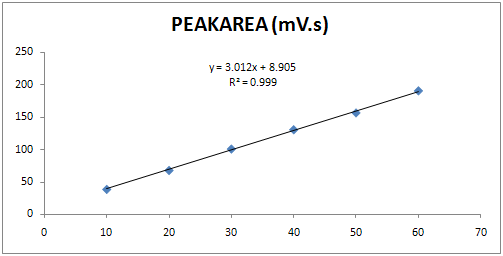
Fig. 3: Calibration curve of Sitagliptin at 260 nm
Linearity and range
Appropriate aliquots of standard Sitagliptin stock solutions (100μg / ml) were taken in different 10 ml volumetric flask andresultant solution was diluted up to the mark with Mobile phase(diluent) to obtain final concentration of 10?60μg / ml. The solutions were preparedin triplicate. Calibration curve were constructed by plotting the concentration of Sitagliptin versus corresponding mean peakarea. The results show that an excellent correlation existsbetweenpeakarea and concentration of drugs within the concentration rangeand the results given in Table 2 and Fig: 3.
Precision
The intra?day precision was determined by analyzing standard solution of concentration 30μg/ml for 6 times on the same daywhile inter?day precision was determined by analyzing corresponding standards daily for 6 day over a period of one week.The values of percentage relative standard deviation (% RSD) forintra?and inter?day variation are given in Table 3.
Accuracy
Accuracy of the method was checked by recovery study usingstandard addition method known amount of standard Sitagliptinwas added into pre analysed sample and subjected it to theproposed high performance liquid chromatographic method. Thesestudies were carried out at three levels i.e,(80, 100 and 120%). Therecovery studies were carried out and the % recovery and standarddeviation of the % recovery were calculated and presented in Table4.
Sensitivity
The sensitivity of measurement of Sitagliptin by use of theproposed method was estimated in terms of the limit of quantitation(LOQ) and the limit of detection (LOD). The LOQ and LOD werecalculated by the use of the equations LOD = 3 x N / B and LOQ = 10x N / B where N is the standard deviation of intercept of calibrationplot and B is the average of the slope of the correspondingcalibration plot. The limit of detection (LOD) was 0.21μg / ml and the limit of quantitation (LOQ) was 0.640μg / ml.
Table 3: Precision results for Sitagliptin
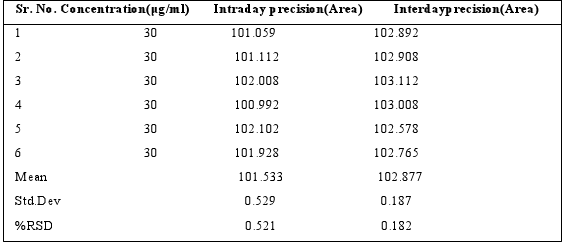
Table 4: Results from recovery studies
|
Brand used |
Label claim (mg/mL) |
Initial amount (µg/mL) |
Amount added (µg/mL) |
Total amount (µg/mL) |
Amount Recovered (µg/mL) |
Recovery |
%RSD |
|
JANUVIA |
100 |
30 30 30
|
24 30 36 |
54 60 66 |
53.998 60.015 66.008 |
99.993 ± 0.23 100.05 ± 0.31 100.02 ± 0.18 |
0.230 0.309 0.179 |
*Average of six determinations
Table 5: Ruggedness studies of Sitagliptin by RPHPLC method
|
Brand used |
Label claim (mg/mL) |
Analyst-I amount found(mg/mL) |
Recovery± SD*(%) |
Analyst-II amount found |
Recovery± SD*(%) |
|
JANUVIA |
100 |
99.84 |
99.20±0.27 |
99.94 |
99.70 ± 0.39 |
*Average of six determinations
Ruggedness and robustness
Ruggedness is a measure of the reproducibility of a test result under normal, expected operating condition from instrument to instrument and from analyst to analyst. The results of ruggedness testing are reported in the Table 5.
Robustness is a measure of capacity of a method to remain unaffected by small but deliberate variations in the method conditions, and is indications of the reliability of the method. Amethod is robust, if it is unaffected by small changes in operating conditions. To determine the robustness of this method, the experimental conditions were deliberately altered at three different
levels and chromatographic response was evaluated. The organic composition in the mobile phase was varied from 55% to 65%, and the variation in mobile phase flow rate by 1.1 ml / min (0.5 and 0.8ml/min) had no significant effect on the retention time and chromatographic response of the 30μg/ml solution, indicating that the method was robust. The results are shown in Table 6.
Table 6: Robustness studies of Sitagliptin by HPLC method
|
Condition |
Modification |
Mean area ± SD* [mV.s] |
RSD (%) |
Mean Rt± SD(min) |
|
Mobile phase composition ( v /v) |
55:45 60:40 65:35 |
101.823 ± 180.68 102.928 ± 160.693 101.958 ± 127.68 |
0.214 0.316 0.485
|
2.21 ± 0.019 2.157 ± 0.015 2.050 ± 0.022 |
|
Mobile phase pH |
3.2 3.5 3.8 |
102.582 ± 163.466 103.002 ± 140.263
101.363 ± 125.362 |
0.382 0.298 0.356
|
2.187 ± 0.014 2.167 ± 0.015 2.20 ± 0.022 |
|
Flow Rate Of Mobile Phase |
1.1 1.2 1.3 |
102.268 ± 136.825 101.865 ± 140.252 102.118 ± 138.568 |
0.269 0.396 0.459 |
2.168 ± 0.02 2.185 ± 0.025 2.21 ±0.02 |
*Average of six determinations
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table 7: Characteristic parameters of Sitagliptin for the RPHPLC method
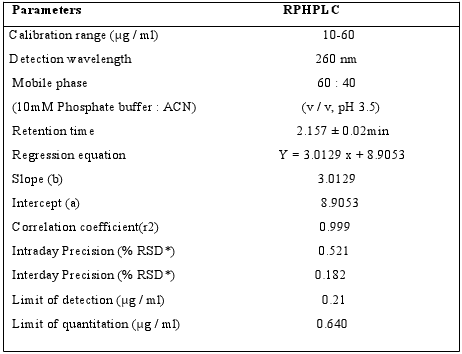
*Y = bx + a, where x is the concentration of compound in μg/ ml and ; Y is the peak area ; *Average of six determinations.
Forced degradation study
When establishing the stability?indicating properties of analytical methods, the intermediate degradation products should not interfere with any stage of drug analysis. The System suitability parameters of forced degradation studies are summarised in Table 8. The results from forced degradation studies are given Table 9.Chromatograms obtained from after degradation under differentstress conditions are shown in Fig: 4 – 8, respectively. No peaks co-elutedwith the drug peak, suggesting the method enabled specificanalysis of Sitagliptin in the presence of its degradationproducts.
Table 8: System suitability parameters of Sitagliptin by degradation studies
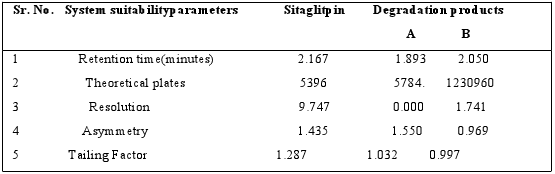
Table 9: Results from analysis of samples by the forced degradation study, showing percentage degradation of Sitagliptin
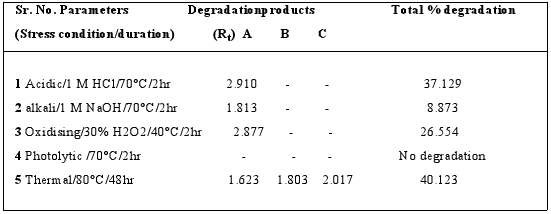
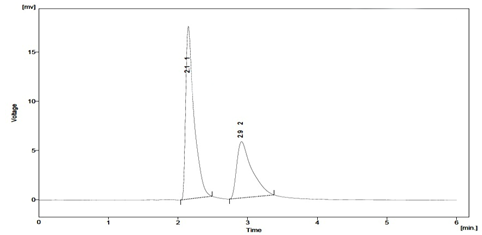
Fig. 4: Typical chromatogram obtained after degradation of Sitaglitpin under acidic conditions
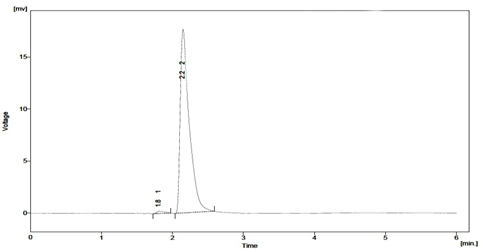
Fig. 5: Typical chromatogram obtained after degradation of Sitagliptin under alkali conditions
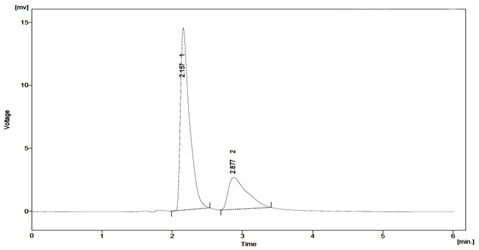
Fig. 6: Typical chromatogram obtained after degradation of Sitagliptin under oxidising conditions
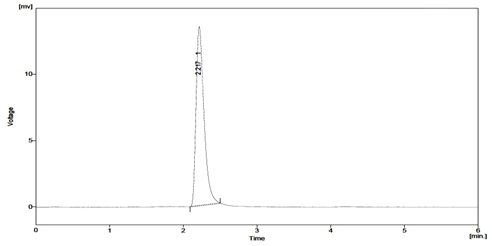
Fig. 7: Typical chromatogram obtained after degradation of Sitagliptin under photolytic conditions
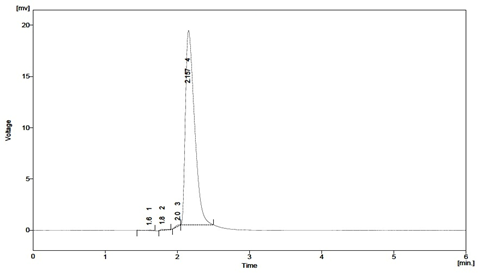
Fig: 8. Typical chromatogram obtained after thermal degradation of Sitagliptin
CONCLUSION
The method developed for quantitative analysis of Sitagliptinis rapid, precise, accurate, and selective. The method was completely validated as per ICH guidelines and satisfactory results were obtained for all the characteristics tested. The method is stability-indicating and can be used to assess the stability of Sitagliptinin bulk and pharmaceutical dosage forms. The method can be conveniently used for assay of Sitagliptin in the bulk drug and in pharmaceutical dosage forms. The method can be conveniently used in quality control laboratory.
ACKNOWLEDGEMENT
We would like thank to Granule chemicals pvt ltd, Hyderabad for providing Pure sample of Sitagliptin and also to the Principal Dr .DevalaRao.Garikapati, K.V.S.R.Siddhartha college of pharmaceutical sciences, Vijayawada for providing facilities to carry work.
REFERENCES
1. Available on drugbank.ca
2. Available on sitgapltin.in
3. Balosekaran.C, Prameela.Rani. A Development and validation of spectrophotometric method for the determination of DPP-4 Inhibitor, Sitagliptin, in its pharmaceutical dosage forms. International Journal of Pharmacy and Pharmaceutical sciences.vol.2,Issue 4,2010.
4. Monika.N, Ravipratap.P, HarshiniShabad. New extractive method development of Sitagliptin Phosphate in API and its Dosage forms by spectrophotometry. International Journal of Pharmacy and Biological sciences.2278-3008vol-1,Issue 6(July-August 2012) Pg:37-40.
5. Swati Kupkar, ShailajaYadhav.Simultaneous estimation of Sitagliptin and Metformin hydrochloride in Bulk and Dosage forms by U.V-Spectrophotometry. International Journal of Pharmacy Research 2012,5(1),580-582.
6. Sheetal Sharma, NimitaManocha, PriyaBhandari. Development of U.V.Spectrophotometry and RP-HPLC method and its validation for simultaneous estimation of Sitagliptin phosphate and simvastatin in marketed formulation. International Journal of Pharmaceutical and Biological Archieves 2012; 3(3):pg:673-678
7. Mohamed Salim, Nehed EI-Enany, FathallahBelah. Simultaneous determination of Sitagliptin and Metformin in pharmaceutical preparations by Capillary Zone electrophoresis and its application to Human plasma analysis. Analytical chemistry Insights 2012:731-46.
8. Phaneemdra.D,V.Venkatesh. Simultaneous estimation of Simvastatin and Sitagliptin by different Analytical methods .International Journals of Advances in Pharmaceutical Analysis.vol-2, Issue 1. 589,No1(2012)
9. ICH, Q2A Text on validation of analytical procedures, Oct, 1994.
10. ICH, Q3B Validation of analytical procedures: methodology,Nov, 1996.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






