
 About Author:
About Author:
Kabita Banik
B.pharm;(WBUT) BCDA College of pharmacy &Technology;
M.pharm(BPUT) Dadhichi college of pharmacy.
banikkabita64@gmail.com
Abstract:
New cancer targeted therapies that make use therapeutic antibodies or small molecules have made treatment more tumor specific and less toxic. Nevertheless, there remain several challenges to the treatment of cancer, including drug resistance, cancer stem cells, and high tumor interstitial fluid pressure. In many solid tumors, for example, increased interstitial fluid pressure makes the uptake of therapeutic agents less efficient. One of the most promising ways of meeting such challenges is ligand-targeted therapy that may be used to make targeting more specific and carry higher dosages of anti-cancer drug to tumor tissue.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1482
Introduction
Antibodies, which initially were viewed as “targeting missiles”, have proved much morecomplex in their targeting and biologic properties than the field’s pioneers envisioned them. MoAbs have emerged as important therapeutic agents for several different malignancies [4]; they have been found to be well-tolerated and effective for the treatment of different cancers, and were consequently approved by theFDA of the US (Table 1). In addition to their own role as anticancer agents, their ability to target tumors also enables them to improve the selectivity of other types of anti-cancer agents, some of which cannot be applied effectively alone.
Murine antibody can be readily transformed into human or humanized formats that are not readily recognized as foreign by the human immune system. In addition, novel antibodybased structures with multiple antigen recognition sites, altered size, or effector domains have been shown to influence the targeting ability of antibodies. Coupled with the identification of appropriate cancer targets, antibodybased therapeutics are finding increasing number of applications in cancer treatment, and they can be effective alone, in conjunction with chemotherapy or radiation therapy, or when conjugated to toxic moieties such as toxins, chemotherapy agents, or radionuclides.
[adsense:468x15:2204050025]
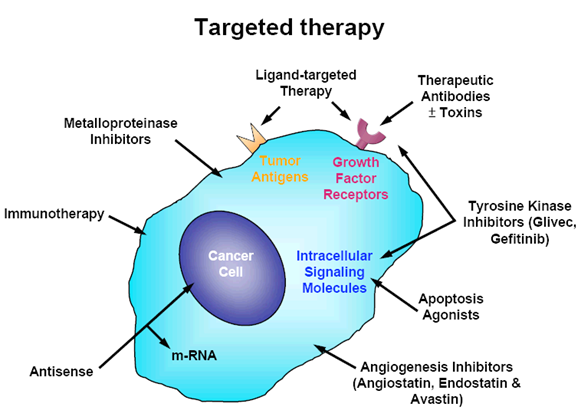
Figure 1:
Targeted therapy refers to a new generation of cancer drugs designed to interfere with a specific target protein that is believed to have a critical role in tumor growth or progression. This approach contrasts with the conventional cytotoxic chemotherapeutics that have been used in major cancer therapy in past decades. The molecular identification of cancer antigens has opened new possibilities for the development of effective immunotherapies, antibodies therapy and ligand-targeted therapy for cancer patients. Ligand-targeted therapy is a successful means of improving the selective toxicity of anticancer therapeutics.
Generation of therapeutic antibodies
The first antibodies studied were murine, rabbit, or rat proteinspurified following immunization of the animal with atarget antigen. Patients often generated antibodies to these foreign antigens; these host antibodies are often referred to as HAMA (human anti-mouse antibody) or HARA (human anti-rat antibody or human anti-rabbit antibody). The host antibody reduces the effectiveness of therapy by prematurely clearing the treatment antibody and limiting the possibilities for future immunotherapy. HAMA or HARA responses can be associated with immune complex-related adverse events such as serum sickness and anaphylaxis. The problem of immunogenicity of murine and chimeric MoAbs could be solved quickly with the progress in MoAb engineering and the generation of fully human antibodies. In solid tumors, the therapeutic agent must overcome several obstacles, including the vascular endothelium, stromal and epithelial barriers and high interstitial pressure [6]. In addition, solid tumors are quite heterogeneous and it is, therefore, difficult to target them completely. When trying to target them, smaller recombinant MoAb structures such as single-chain antibodies should be able to penetrate into the tumor with higher efficacy than the parental antibody [7]. However, this advantage is accompanied by the disadvantage that small structures such as these are more rapidly cleared from the plasma, and therefore they have shorter half-lives [8]. One promising approach to solid tumors is to target the tumor microenvironment in general and the endothelium of tumor blood vessels in particular [9], because several tumor endothelial markers are well characterized [10]. Murine, rabbit, and rat antibodies are not always able to recruit human immune effector functions, such as antibodydependent cell-mediated cytotoxicity (ADCC) and complement- dependent cytotoxicity (CDC), which are needed to facilitate destruction of a malignant cell.
Monocytes, macrophages, natural killer (NK) cells, killer cells, and granulocytes express Fc receptors and can exert cytotoxic effects. ADCC results when the bound antibody binds NK cells. The Fab portion of the antibody attaches to the malignant cell while the opposing Fc region binds to Fc receptors on NK cells. The NK cells in turn release cell-lysing molecules that destroy the target. Genetically engineered antibodies have also enhanced efficacies through longer half-life. The half-life of the chimeric anti-CD20 antibody, rituximab, is 76 h after a single infusion and 206 h after four infusions, compared with 28 h for the murine counterpart, ibritumomab [12,13].
Origins of monoclonal antibody therapy
Monoclonal antibodies for cancer
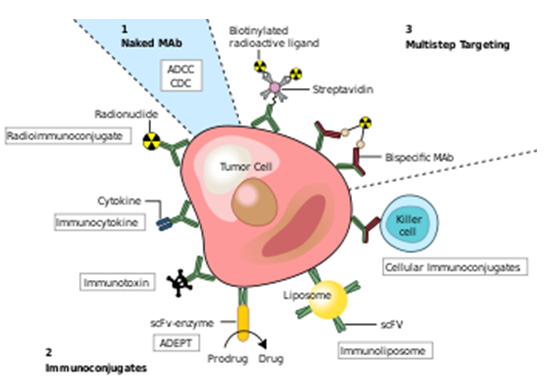
Fig:2
Monoclonal antibodies for cancer. ADEPT, antibody directed enzyme prodrug therapy; ADCC, antibody dependent cell-mediated cytotoxicity; CDC, complement dependent cytotoxicity; MAb, monoclonal antibody; scFv, single-chain Fv fragment.[4]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Immunotherapy developed as a technique with the discovery of the structure of antibodies and the development of hybridoma technology, which provided the first reliable source of monoclonal antibodies.[5] These advances allowed for the specific targeting of tumors both in vitro and in vivo. Initial research on malignant neoplasms found MAb therapy of limited and generally short-lived success with malignancies of the blood.[6][7] Furthermore treatment had to be specifically tailored to each individual patient, thus proving to be impracticable for the routine clinical setting.
Throughout the progression of monoclonal drug development there have been four major antibody types developed: murine, chimeric, humanised and human.
Initial therapeutic antibodies were simple murine analogues, which contributed to the early lack of success. It has since been shown that these antibodies have: a short half-life in vivo (due to immune complex formation), limited penetration into tumour sites, and that they inadequately recruit host effector functions.[8] To overcome these difficulties the technical issues initially experienced had to be surpassed. Chimeric and humanized antibodies have generally replaced murine antibodies in modern therapeutic antibody applications. Hybridoma technology has been replaced by recombinant DNA technology, transgenic mice and phage display.[9] Understanding of proteomics has proven essential in identifying novel tumour targets.
Murine monoclonal antibodies (suffix -omab)
Initially, murineantibodies were obtained by hybridoma technology, for which Kohler and Milstein received a Nobel prize. However the dissimilarity between murine and human immune systems led to the clinical failure of these antibodies, except in some specific circumstances. Major problems associated with murine antibodies included reduced stimulation of cytotoxicityand the formation complexes after repeated administration, which resulted in mild allergic reactionsand sometimes anaphylactic shock.[8]
Chimeric and humanized monoclonal antibodies (suffixes -ximab, -zumab respectively)
To reduce murine antibody immunogenicity, murine molecules were engineered to remove immunogenic content and to increase their immunologic efficiency.[8]This was initially achieved by the production of chimericand humanized antibodies. Chimeric antibodies are composed of murine variable regions fused onto human constant regions. Human gene sequences, taken from the kappa light chain and the IgG1 heavy chain, results in antibodies that are approximately 65% human. This reduces immunogenicity, and thus increases serumhalf-life.
Humanised antibodies are produced by grafting murine hypervariable [disambiguation needed]amino acid domains into human antibodies. This results in a molecule of approximately 95% human origin. However it has been shown in several studies that humanised antibodies bind antigen much more weakly than the parent murine monoclonal antibody, with reported decreases in affinity of up to several hundredfold.[10][11]Increases in antibody-antigen binding strength have been achieved by introducing mutationsinto the complementarity determining regions(CDR),[12]using techniques such as chain-shuffling, randomization of complementarity determining regions and generation of antibody libraries with mutations within the variable regions by error-prone PCR, E. colimutator strains, and site-specific mutagenesis.[1]
Human monoclonal antibodies (suffix -umab)
Human monoclonal antibodies are produced using transgenicmice or phage displaylibraries. Human monoclonal antibodies are produced by transferring human immunoglobulin genes into the murine genome, after which the transgenic mouse is vaccinatedagainst the desired antigen, leading to the production of monoclonal antibodies.,[9]allowing the transformation of murine antibodies in vitro into fully human antibodies.[3]
The heavy and light chains of human IgG proteins are expressed in structural polymorphic (allotypic) forms. Human IgG allotype has been considered as one of the many factors that can contribute to immunogenecity.[13]The general scheme of a monoclonal antibody development program is described in.[14]
Targeted conditions
Cancer
Anti-cancer monoclonal antibodies can be targeted against malignant cells by several mechanisms:
- Radioimmunotherapy(RIT) involves the use of radioactivelyconjugated murine antibodies against cellular antigens. Most research currently involved their application to lymphomas, as these are highly radio-sensitive malignancies. To limit radiation exposure, murine antibodies were especially chosen, as their high immunogenicity promotes rapid clearance from the body. Tositumomabis an exemplar used for non-Hodgkins lymphoma.
- Antibody-directed enzyme prodrug therapy (ADEPT) involves the application of cancer associated monoclonal antibodies which are linked to a drug-activating enzyme. Subsequent systemic administration of a non-toxic agent results in its conversion to a toxic drug, and resulting in a cytotoxic effect which can be targeted at malignant cells. The clinical success of ADEPT treatments has been limited to date.[15]However it holds great promise, and recent reports suggest that it will have a role in future oncological treatment.
- Immunoliposomesare antibody-conjugated liposomes. Liposomes can carry drugs or therapeutic nucleotidesand when conjugated with monoclonal antibodies, may be directed against malignant cells. Although this technique is still in its infancy, significant advances have been made. Immunoliposomes have been successfully used in vivo to achieve targeted delivery of tumour-suppressing genes into tumours, using an antibody fragment against the human transferrinreceptor. Tissue-specific gene delivery using immunoliposomes has also been achieved in brain, and breast cancer tissue.[16]
Example FDAapproved therapeutic monoclonal antibodies[1]
FDA approved therapeutic antibodies
For a more comprehensive list, see List of monoclonal antibodies.
The first FDA-approved therapeutic monoclonal antibody was a murine IgG2a CD3 specific transplant rejectiondrug, OKT3(also called muromonab), in 1986. This drug found use in solid organ transplantrecipients who became steroidresistant.[18]Hundreds of therapies are undergoing clinical trials. Most are concerned with immunological and oncological targets.
Table:1
|
Example FDAapproved therapeutic monoclonal antibodies[1] |
||||||
|
Antibody |
Brand name |
Company |
Approval date |
Type |
Target |
Indication |
|
|
|
|
|
|
|
|
|
Alemtuzumab |
Campath |
Genzyme |
2001 |
humanized |
CD52 |
Chronic lymphocytic leukemia |
|
|
|
|
|
|
|
|
|
Bevacizumab |
Avastin |
Genentech/Roche |
2004 |
humanized |
Vascular endothelial growth factor (VEGF) |
Colorectal cancer, Age related macular degeneration (off-label) |
|
Brentuximab vedotin |
Adcetris |
|
2011 |
Chimeric |
CD30 |
Anaplastic large cell lymphoma(ALCL) and Hodgkin lymphoma |
|
Canakinumab |
Ilaris |
Novartis |
2009 |
Human |
IL-1β |
Cryopyrin-associated periodic syndrome(CAPS) |
|
Cetuximab |
Erbitux |
Bristol-Myers Squibb/Eli Lilly/Merck KGaA |
2004 |
chimeric |
epidermal growth factor receptor |
Colorectal cancer, Head and neck cancer |
|
|
|
|
|
|
|
|
|
Gemtuzumab |
Mylotarg |
Wyeth |
2000 |
humanized |
CD33 |
Acute myelogenous leukemia(with calicheamicin) |
|
|
|
|
|
|
|
|
|
Ibritumomab tiuxetan |
Zevalin |
Spectrum Pharmaceuticals, Inc. |
2002 |
murine |
CD20 |
Non-Hodgkin lymphoma(with yttrium-90or indium-111) |
|
Ipilimumab( MDX-101 ) |
Yervoy |
|
2011 |
Human |
blocks CTLA-4 |
Melanoma |
|
|
|
|
|
|
|
|
|
Ofatumumab |
Arzerra |
|
2009 |
Human |
CD20 |
Chronic lymphocytic leukemia |
|
|
|
|
|
|
|
|
|
Panitumumab |
Vectibix |
Amgen |
2006 |
human |
epidermal growth factor receptor |
Colorectal cancer |
|
|
|
|
|
|
|
|
|
Rituximab |
Rituxan, Mabthera |
Biogen Idec/Genentech |
1997 |
chimeric |
CD20 |
Non-Hodgkin lymphoma |
|
|
|
|
|
|
|
|
|
Tositumomab |
Bexxar |
GlaxoSmithKline |
2003 |
murine |
CD20 |
Non-Hodgkin lymphoma |
|
Trastuzumab |
Herceptin |
Genentech |
1998 |
humanized |
ErbB2 |
Breast cancer |
Targeted therapy by small molecules
The growing understanding of the molecular events underlying the etiology of different cancers as well as the signalling events that are critical for the continued growth and proliferation of cancer cells has enhanced the opportunities to develop novel agents. This new type of chemotherapy has been termed targeted therapy, and the goal of this modern chemotherapy is to provide molecular levels-based agents that are more specific for cancer cells. In most current drug discovery programs, rational and empiric approaches are being used either in parallel or in combination with one another. Lead compounds are identified as inhibitors for molecular targets through molecular screening [45]. Protein phosphorylation regulates most aspects of cell life, whereas abnormal phosphorylation is a cause or consequence of disease especially in cancer biology, such as abnormal proliferation, anti-apoptosis and angiogenesis [46-48]. Many studies have also indicated that activation of protein phosphorylation-related pathways in tumors can occur through mutation or overexpression if compared to normal cells [49,50]. For these reasons, the targeted therapeutics ascribes to pharmacological agents that are as close to be mono-specific as possible to avoid the detrimental side effects that sometimes occur with traditional therapies. Therefore,small molecule inhibitors of protein kinases have emerged as indispensable for studying target therapy [51]. The protein kinases that have been targeted most intensively for drug development are plasma membrane-associated protein tyrosine kinases [52]. The first kinase inhibitors were described nearly 20 years ago and developed in the early 1980s by Hiroyoshi Hidaka. Naphthalene sulphonamides had already been developed as antagonists of the calcium-binding protein calmodulin [53,54]. There are more than 30 such agents in clinical trials now [55] and the most well-known small molecule inhibitors are glivec and gefitinib (Figure 2). Glivec (imitanib mesylate, Gleevec, STI571; Novartis) is the first selective tyrosine kinase inhibitor to be approved for the treatment of cancer in 2001 [56]. Glivec is a 2- phenylaminopyrimidine which competitively inhibits ATP binding to the Abl kinase, thereby inhibiting the constitutively activated Bcr-Abl tyrosine kinase, which is a specific genetic change encoding abnormal protein associated with human cancer [57-59] (Figure 2A). As the tyrosine kinase activity of Bcr–Abl is crucial for its transforming activity, the enzymatic activity of this deregulated gene could plausibly be defined as an attractive drug target for addressing Bcr- Abl-related chronic myelogenous leukaemia (CML).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
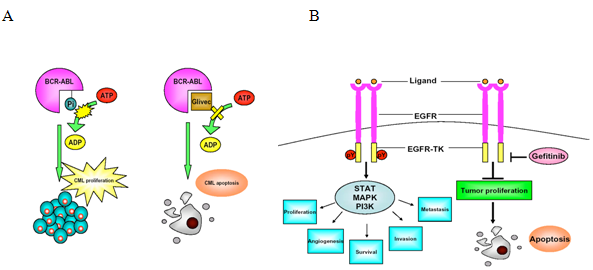
Figure 3: Mechanisms of small molecule inhibitors for tumor treatment. Most of the small molecule inhibitors are designed to specifically target overexpressed or mutated signaling pathway in tumor cells rather than normal cells. And then, the life cycle of tumor cells will be blocked and triggered to apoptosis. (A) Constitutively activated Bcr-Abl tyrosine kinase causes chronic myelogenous leukaemia (CML). The activity of Bcr-Abl is catalyzed by ATP, and the phosphorylation binding site can be inhibited by glivec. Therefore, the tumor cells’ proliferation will be terminated and alternatively switch to apoptosis pathway. (B) EGFR-mediated signaling contributes to the up-regulation of many processes that are essential for tumor growth and progression. Gefitinib is a small molecule that inhibits ATP binding within the tyrosine kinase domain of EGFR, which inhibitsEGFR autophosphorylation and consequently blocked signal transduction from activated EGFR. The critical mechanisms of tumor growth are inhibited as following the gefitinib treatment.
Ligand-targeted therapy
Most cancer cells share many common features with the normal host cells from which they are derived. Therefore, high levels of selective toxicity cannot be achieved with anticancer chemotherapeutics because of the lack of unique molecular targets that would distinguish them from normal cells. This can lead to increased toxicities against normal tissues, including bone marrow, gastrointestinal tract and hair follicle tissues. Furthermore, trying to avoid the side effects that occur as a result of toxicities to normal tissues, we often give sub-optimal doses of anticancer chemotherapeutics, resulting in the eventual failure of therapy, which is often accompanied by the development of drug resistance and metastatic disease. The selective toxicity of an anticancer drug can be increased by either increasing the amount of the drug that reaches the cancer tissue or by decreasing the concentration of drug that reaches at the normal tissues. Therefore, ligand-targeted therapy makes possible tumor specificity and limited toxicity and shows promise in the development of novel therapies for cancer. Ligand-targeted therapy can carry higher doses of a drug to the tumor tissue and may overcome obstacles presented by cytotoxic chemotherapy. There are several obstacles in cancer therapy including drug resistance, high tumor interstitial fluid pressure (IFP), and cancer stem cells (CSCs).
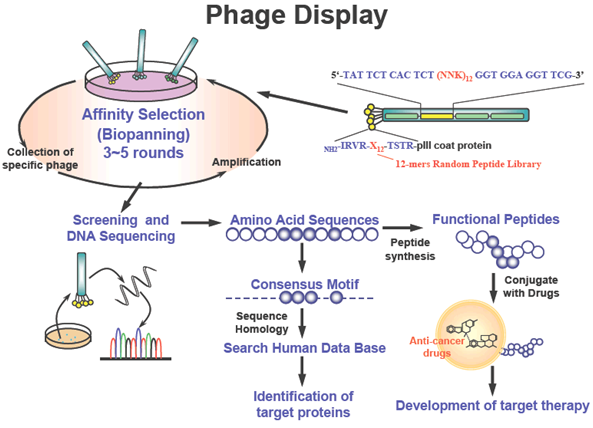
Figure 4: Identification of targeting ligands to cancer cells by phage display. Peptide or antibody libraries can be expressed as fusion proteins with a coat protein (pIII) of a bacteriophage, resulting in display of fused proteins on the surface of virion. Affinity selection (biopanning) of phagedisplayed peptide libraries represents a powerful means of identifying peptide ligands for targets of interest. For screening of targeting ligands, phage-displayed peptide library was pre-cleared by normal cells and affinity selection with cancer cells. After biopanning three to five times, targeting phage clones were selected by ELISA, flow cytometry, immunofluorescence, and in vivo homing assays. Targeting ligands were further identified and characterized by synthetic peptide binding and competition assay. Targeting ligands can be used to identify cell surface markers and develop ligand-targeted therapy.
Search targeting ligands using phage display Phage display, a selection technique in which a peptide or protein is expressed as a fusion with a coat protein of bacteriophage, results in a display of the fusion peptide or protein on the surface of the virion. Phage-displayed random peptide libraries provide opportunities to map B-cell epitopes [10-14] and protein–protein contacts [15-18], select bioactive peptides bound to receptors [19-12] or proteins [16,22-25], search for disease-specific antigen mimics [12-12], and determine cell- [29-31] and organ-specific peptides [12,32-14]. Recently, we have developed phage display methods to identify the receptors expressed specifically on cancer cells and tumor vessels. The strategy for identification of tumortargeting ligand is shown in Figure 6. Using these technologies, we identified a 12-mer peptide (L-peptide) specifically binding to nasopharyngeal carcinoma (NPC) cells. The Lphage and synthetic L-peptide bound to the tumor cell surfaces of most NPC cell lines and biopsy specimens [11]. In SCID mice bearing NPC xenograft, the L-phages specifically bound to the tumor mass. Synthetic L-peptide has been shown to inhibit the binding of L-phage particles to the tumor mass in the competitive inhibition assay [131]. Once we have discovered the targeting ligands to the cancer cells, we can conjugate the targeting peptides with chemotherapeutic drugs and develop ligand-targeted therapies to kill cancer cells specifically. We have chosen liposomes to conjugate with targeting ligands because of the following advantages:
(i) prolonged blood circulation,
(ii) sufficient tumor accumulation,
and (iii) controlled drug release and uptake by tumor cells with a release profile matching the pharmacodynamics of the drug.
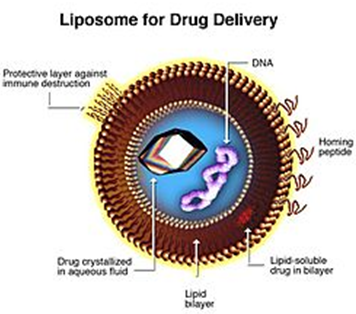
Fig 5- Liposomes are composite structures made of phospholipids and may contain small amounts of other molecules. Though liposomes can vary in size from low micrometer range to tens of micrometers, unilamellar liposomes, as pictured here, are typically in the lower size range with various targeting ligands attached to their surface allowing for their surface-attachment and accumulation in pathological areas for treatment of disease.[1]
Targeting cancer (liposomes)
New Liposomal drugs targeting cancer like Liposomal Cisplatin (Lipoplatin of Regulon Inc.) has received Orphan Drug designation for Pancreatic Cancer from EMEA.[citation needed
Conclusion
Pharmaceutical industry has been successful in discovering many new cytotoxic drugs that can potentially be used for the treatment of cancer, this life-threatening disease still causes near 7 million deaths every year worldwide and the number is growing.
Thus, the ongoing obligation to the design and discovery of new cancer therapy is urgent. In general, cancer chemotherapy is usually accompanied by severe side effects and acquired drug resistance. Therefore, we anxiously await the development of target therapy that will allow greater targeting.
Recently, some attempts have been made for this purpose including the usage of monoclonal antibodies [20,22,41] or small molecules [56,82] to inhibit the tumor growth. Despite the promising clinical results from the agents that we have highlighted, there is still significant limitation to the concept of “pathwayspecific” targeted therapies. These agents are only effective in tumor types that are dependent upon the tumor antigens that are expressed or the pathways that are being inhibited. It is readily apparent that most solid tumors are the result of numerous genetic mutations, and thus inhibiting a single cellular pathway may not result in a significant therapeutic outcome. Design of agents that target a number of pathways will possibly increase the therapeutic effect, but also increase the risk of treatment-related toxicities. Liposomes containing various lipid derivatives of polyethylene glycol (PEG) have resulted in extension of the half life [15]. However, they need a tumor targeting ligand to carry them to the tumor site. For solid malignancies, which comprise more than 90% of human cancers, antibodies recognizing tumorspecific antigens have provided only some utility for drug delivery because the immunoconjugates cannot easily penetratethe tumor tissue [17,15]. Therefore, identification of peptide ligands and development of peptide-targeting
liposome is highly desirable. Ligand-targeted therapy via targeting liposome may be able to allow us to carry higher dosage of drugs to the tumor tissue and help us overcome some of the obstacles to effective cancer therapy.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
References
1. Papac RJ. Origins of cancer therapy. Yale J Biol Med 74: 391- 398, 2001.
2. Dunn FB. National Cancer Act: leaders reflect on 30 years of progress. J Natl Cancer Inst 94: 8-9, 2002.
3. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256: 495-497, 1975.
4. Harris M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol 5: 292-302, 2004. 5. Parker BA, Vassos AB, Halpern SE, Miller RA, Hupf H, Amox DG, Simoni JL, Starr RJ, Green MR, Royston I. Radioimmunotherapy of human B-cell lymphoma with 90Y-conjugated antiidiotype monoclonal antibody. Cancer Res 50: 1022s-1028s, 1990.
6. Stohrer M, Boucher Y, Stangassinger M, Jain RK. Oncotic pressure in solid tumors is elevated. Cancer Res 60: 4251-4255, 2000.
7. Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res 52: 3402-3408, 1992.
8. Adams GP, Schier R, Marshall K, Wolf EJ, McCall AM, Marks JD, Weiner LM. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res 58: 485-490, 1998.
9. Hofmeister V, Vetter C, Schrama D, Brocker EB Becker JC. Tumor stroma-associated antigens for anti-cancer immunotherapy. Cancer Immunol Immunother 55: 481-494, 2006.
10. Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer 5: 436-446, 2005.
11. Newman R, Alberts J, Anderson D, Carner K, Heard C, Norton F, Raab R, Reff M, Shuey S, Hanna N. "Primatization" of recombinant antibodies for immunotherapy of human diseases: a macaque/ human chimeric antibody against human CD4. Biotechnology 10: 1455-1460, 1992.
12. McLaughlin P, Grillo-Lopez AJ, Link BK., Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a fourdose treatment program. J Clin Oncol 16: 2825-2833, 1998.
13. Witzig TE, White CA, Wiseman GA, Gordon LI, Emmanouilides C, Raubitschek A, Janakiraman N, Gutheil J, Schilder RJ, Spies S, Silverman DH, Parker E, Grillo-Lopez AJ. Phase I/II trial of IDECY2B8 radioimmunotherapy for treatment of relapsed or refractory CD20(+) B-cell non-Hodgkin's lymphoma. J Clin Oncol 17: 3793-3803, 1999.
14. Liu AY, Robinson RR, Hellstrom KE, Murray ED Jr, Chang C P, Hellstrom I. Chimeric mouse-human IgG1 antibody that can mediate lysis of cancer cells. Proc Natl Acad Sci USA 84: 3439-3443, 1987.
15. Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature 332: 323-327, 1988.
16. Grillo-Lopez AJ, Hedrick E, Rashford M, Benyunes M. Rituximab: ongoing and future clinical development. Semin Oncol 29: 105-112, 2002.
17. Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol Cell Biol 9: 1165-1172, 1989.
18. Slamon D, Pegram M. Rationale for trastuzumab (Herceptin) in adjuvant breast cancer trials. Semin Oncol 28: 13-19, 2001.
19. Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA 89: 4285-4289, 1992.
20. Gerber HP, Ferrara N. The role of VEGF in normal and neoplastic hematopoiesis. J Mol Med 81: 20-31, 2003.
21. Press OW, Appelbaum F, Ledbetter JA, Martin PJ, Zarling J, Kidd P, Thomas ED. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood 69: 584-591, 1987.
22. Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, Hanna N, Anderson DR. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 83: 435-445, 1994.
23. Maloney DG, Liles TM, Czerwinski DK Waldichuk C, Rosenberg J, Grillo-Lopez A, Levy R. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood 84: 2457-2466, 1994.
24. Czuczman MS, Grillo-Lopez AJ, White CA, Saleh M, Gordon L, LoBuglio AF, Jonas C, Klippenstein D, Dallaire B, Varns C. Treatment of patients with low-grade B-cell lymphoma with the combination of chimeric anti-CD20 monoclonal antibody and CHOP chemotherapy. J Clin Oncol 17: 268-276, 1999.
25. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire W.L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177- 182, 1987.
26. Naito K, Takeshita A, Shigeno K, Nakamura S, Fujisawa S, Shinjo K, Yoshida H, Ohnishi K, Mori M, Terakawa S, Ohno R. Calicheamicin-conjugated humanized anti-CD33 monoclonal antibody (gemtuzumab zogamicin, CMA-676) shows cytocidal effect on CD33-positive leukemia cell lines, but is inactive on Pglycoprotein- expressing sublines. Leukemia 14: 1436-1443, 2000.
27. Boghaert ER, Khandke K, Sridharan L, Armellino D, Dougher M, Dijoseph JF, Kunz A, Hamann PR, Sridharan A, Jones S, Discafani C, Damle NK. Tumoricidal effect of calicheamicin immuno- conjugates using a passive targeting strategy. Int J Oncol 28: 675-684, 2006.
28. Genentech Inc. Herceptin® (trastuzumab). in: Investigator brochure, Sept. San Francisco, 1998.
29. Lozanski G, Heerema NA, Flinn IW, Smith L, Harbison J, Webb J, Moran M, Lucas M, Lin T, Hackbarth ML, Proffitt JH, Lucas D, Grever MR, Byrd JC. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 103: 3278-3281, 2004.
30. Kaufman DB, Leventhal JR, Gallon LG, Parker MA. Alemtuzumab induction and prednisone-free maintenance immunotherapy in simultaneous pancreas-kidney transplantation comparison with rabbit antithymocyte globulin induction longterm results. Am J Transplant 6: 331-339, 2006.
31. Lundin J, Osterborg A, Brittinger G, Crowther D, Dombret H, Engert A, Epenetos A, Gisselbrecht C, Huhn D, Jaeger U, Thomas J, Marcus R, Nissen N, Poynton C, Rankin E, Stahel R, Uppenkamp M, Willemze R, Mellstedt H. CAMPATH-1H monoclonal antibody in therapy for previously treated low-grade non- Hodgkin's lymphomas: a phase II multicenter study. European Study Group of CAMPATH-1H Treatment in Low-Grade Non- Hodgkin's Lymphoma. J Clin Oncol 16: 3257-3263, 1998.
32. Chinn PC, Leonard JE, Rosenberg J, Hanna N, Anderson DR. Preclinical evaluation of 90Y-labeled anti-CD20 monoclonal antibody for treatment of non-Hodgkin's lymphoma. Int J Oncol 15: 1017-1025, 1999. Wu et al. J. Cancer Mol. 2(2): 57-66, 2006 64 Print ISSN 1816-0735; Online ISSN 1817-4256
33. Cheson BD. Radioimmunotherapy of non-Hodgkin lymphomas. Blood 101: 391-398, 2003.
34. Nademanee A, Forman S, Molina A, Fung H, Smith D, Dagis A, Kwok C, Yamauchi D, Anderson AL, Falk P, Krishnan A. Kirschbaum M, Kogut N, Nakamura R, O'Donnell, , Parker P, Popplewell L, Pullarkat V, Rodriguez R, Sahebi F, Smith E, Snyder D, Stein A, Spielberger R, Zain J, White C, Raubitschek A. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkin lymphoma. Blood 106: 2896- 2902, 2005.
35. Kaminski MS, Zasadny KR, Francis IR, Fenner MC, Ross CW, Milik AW, Estes J, Tuck M, Regan D, Fisher S, Glenn SD, Wahl RL. Iodine-131-anti-B1 radioimmunotherapy for B-cell lymphoma. J Clin Oncol 14: 1974-1981, 1996.
36. Vose JM, Wahl RL, Saleh M, Rohatiner AZ, Knox SJ, Radford JA, Zelenetz AD, Tidmarsh GF, Stagg RJ, Kaminski MS. Multicenter phase II study of iodine-131 tositumomab for chemotherapyrelapsed/ refractory low-grade and transformed low-grade B-cell non-Hodgkin's lymphomas. J Clin Oncol 18: 1316-1323, 2000.
37. Liu SY, Eary JF, Petersdorf SH, Martin PJ, Maloney DG, Appelbaum FR, Matthews DC, Bush SA, Durack LD, Fisher DR, Gooley TA, Bernstein ID, Press OW. Follow-up of relapsed B-cell lymphoma patients treated with iodine-131-labeled anti-CD20 antibody and autologous stem-cell rescue. J Clin Oncol 16: 3270- 3278, 1998.
38. Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res 59: 1935-1940, 1999.
39. Baselga J, Pfister D, Cooper MR, Cohen R, Burtness B, Bos M, D'Andrea G, Seidman A, Norton L, Gunnett K, Falcey J, Anderson V, Waksal H, Mendelsohn J. Phase I studies of antiepidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J Clin Oncol 18: 904-914, 2000.
40. Jimeno A, Rubio-Viqueira B, Amador ML, Oppenheimer D, Bouraoud N, Kulesza P, Sebastiani V, Maitra A, Hidalgo M. Epidermal growth factor receptor dynamics influences response to epidermal growth factor receptor targeted agents. Cancer Res 65: 3003-3010, 2005.
41. Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, Chung DC, Sahani DV, Kalva SP, Kozin SV, Mino M, Cohen KS, Scadden DT, Hartford AC, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Chen HX, Shellito PC, Lauwers GY, Jain RK. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 10: 145-147, 2004.
42. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3: 391-400, 2004.
43. Sonpavde G. Bevacizumab in colorectal cancer. N Engl J Med 351: 1690-1691, 2004.
44. Kabbinavar FF, Schulz J, McCleod M, Patel T, Hamm JT, Hecht JR, Mass R, Perrou B, Nelson B, Novotny WF. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. J Clin Oncol 23: 3697-3705, 2005.
45. Chu E. Drug development. in: Vincent T, DeVita J, Hellman S, Rosenberg SA, ed. Cancer: Principles and Practice of Oncology, Philadelphia: Lippincott Williams and Wilkin 2005.
46. Cicenas J, Urban P, Kung W, Vuaroqueaux V, Labuhn M, Wight E, Eppenberger U, Eppenberger-Castori S. Phosphorylation of tyrosine 1248-ERBB2 measured by chemiluminescence-linked immunoassay is an independent predictor of poor prognosis in primary breast cancer patients. Eur J Cancer 42: 636-645, 2006.
47. Maatta JA, Sundvall M, Junttila TT, Peri L, Laine VJ, Isola J, Egeblad M, Elenius K. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligandindependent survival and cancer cell growth. Mol Biol Cell 17: 67-79, 2006.
48. Troussard AA, McDonald PC, Wederell ED, Mawji NM, Filipenko NR, Gelmon KA, Kucab JE, Dunn SE, Emerman JT, Bally MB, Dedhar S. Preferential dependence of breast cancer cells versus normal cells on integrin-linked kinase for protein kinase B/Akt activation and cell survival. Cancer Res 66: 393-403, 2006.
49. Freedman NJ, Kim LK, Murray JP, Exum ST, Brian L, Wu JH, Peppel K. Phosphorylation of the platelet-derived growth factor receptor-beta and epidermal growth factor receptor by G protein- coupled receptor kinase-2. Mechanisms for selectivity of desensitization. J Biol Chem 277: 48261-48269, 2002.
50. Lammering G, Hewit TH, Hawkins WT, Contessa JN, Reardon DB, Lin PS, Valerie K, Dent P, Mikkelsen RB, Schmidt-Ullrich RK. Epidermal growth factor receptor as a genetic therapy target for carcinoma cell radiosensitization. J Natl Cancer Inst 93: 921-929, 2001.
51. Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol 12: 621-637, 2005.
52. Cohen P. The development and therapeutic potential of protein kinase inhibitors. Curr Opin Chem Biol 3: 459-465, 1999.
53. Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry 23: 5036-5041, 1984.
54. Cohen P. Protein kinases--the major drug targets of the twentyfirst century? Nat Rev Drug Discov 1: 309-315, 2002.
55. Dancey J, Sausville EA. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov 2: 296-313, 2003.
56. Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 1: 493-502, 2002.
57. Clark J, Cools J, Gilliland DG. EGFR inhibition in non-small cell lung cancer: resistance, once again, rears its ugly head. PLoS Med 2: e75, 2005.
58. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 344: 1038-1042, 2001.
59. Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW, Fischer T, O'Brien SG, Stone RM, Gambacorti-Passerini CB, Russell NH, Reiffers JJ, Shea TC, Chapuis B, Coutre S, Tura S, Morra E, Larson RA, Saven A, Peschel C, Gratwohl A, Mandelli F, Ben-Am M, Gathmann I, Capdeville R, Paquette RL, Druker BJ. Imatinib induces hematologic and cyt








