

 About Authors:
About Authors:
Ajay Kumar*1, Dr. Bharat Prashar2, Robin Sharma1
1M. Pharm (Pharmacology)
2Head of Pharmacy Department
Manav Bharti University, Solan.
*ajaykumarsharma423@hotmail.com
ABSTRACT
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, primarily manifesting as a loss of memory in old aged people. In this disease the destruction of neurons in the cortex and limbic structures of the CNS occurs, particularly in basal forebrain, amygdala, hippocampus, cerebral cortex. These areas are associated with functions of higher learning, memory, reasoning, behaviour, identification, an emotional control in brain. Alzheimer's disease is a devastating disease whose recent increase in incidence rates has broad implications for rising health care costs. Clinically, patients initially present with short-term memory loss, subsequently followed by executive dysfunction, confusion, agitation, and behavioural disturbances. This review comprises aspects of the introduction, history, types, etiology, pathophysiologic mechanisms of neurological pathways of Alzheimer’s disease and the drug treatments and their target site.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1448
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease characterized by progressive cognitive decline, including memory loss, behavioral and psychological symptoms and personality changes. The neuropathological hallmarks of AD are the presence of neuritic (senile) plaques (NP) and neurofibrillary tangles (NFT), along with neuronal loss, dystrophic neurites, and gliosis. Neuritic plaques are extracellular lesions and their main constituent is the amyloid-β42 peptide (Aβ42). Neurofibrillary tangles are intracellular lesions that are mainly composed of hyperphosphorylated Tau protein.
Alzheimer's disease (AD) is a progressive neurodegenerative disease that accounts for the majority (50 to 75%) of dementia cases in clinical and population samples [1, 2] .The clinical manifestations of AD include the progressive deterioration of episodic memory and other intellectual abilities, leading to global cognitive decline, behavioral and psychiatric symptoms and personality changes. Most AD cases begin after 65 years old (i.e. late-onset AD). However, some cases manifest in younger subjects (i.e. early-onset AD). The dementia syndrome in these patients usually begins in the 5th or 6th decade of life.
Age and low-educational status are the most important risk factors for late-onset AD[3] . Other important risk factors include the presence of the allele ?4 of the apolipoprotein E (APOE) gene, history of head trauma with loss of consciousness, life-long uncontrolled cardiovascular risk factors (hypertension, diabetes mellitus, dyslipidemia), sedentary life style, life-long low cognitive demand [4]. On the other hand, early-onset AD is usually associated to genetic mutations, the most commonly described being the Amyloid Precursor Protein (APP) gene on chromosome 21, and the Presenilin 1 and 2 genes on chromosomes 14 [5].
Regardless of age of dementia onset, the neuropathological findings in AD patients are very similar. The hallmarks of AD are the presence of senile (neuritic) plaques (NP) and neurofibrillary tangles (NFT), together with neuronal loss, dystrophic neurites, and gliosis [6]. Neuritic plaques are extracellular lesions and their main constituent is the amyloid-β42 peptide (Aβ42). Neurofibrillary tangles are intracellular lesions and are mostly composed of hyperphosphorylated TAU protein. Despite controversial findings, the progression of the clinical AD dementia syndrome correlates with the pattern of progression of these lesions in the brain [7].
[adsense:468x15:2204050025]
History
AD is identified by German physician, Dr. Alois Alzheimer in the year 1906, specifically identified a collections of abnormalities in the brain cells .One of Dr. Alzheimer’s patients died after years of severe memory problems, confusion and difficulty understanding questions. Upon the death death of that patient, while performing a brain autopsy, he noted dense deposits surrounding the nerve cells (neuritic plaques) and inside the nerve cells he observed twisted bands of fibers (neurofibrillary tangles). Today, this degenerative brain disorders bears his name, and when found during an autopsy, these plaques and tangles mean a definite diagnosis of AD.
The scientists discovered another link between cognitive decline and the number of plaques and tangles in the brain in the year 1960. The medical community then formally recognized Alzheimer’s as a disease and not a normal part of aging. This attention get increased when the important discoveries and a better understanding of complex nerve cells in the brains of AD patients is led down in 1990. More research was done on AD susceptibility genes, and several drugs were approved to treat the cognitive symptoms of the disease [8].
Types of AD
Presenile or early onset Alzheimer’s disease
Researchers have directed their energies towards understanding very early stage AD, reasoning that any defects appearing early are likely to be more relevant to the true underlying cause. These efforts have borne fruit in at least two related directions.
• Researchers have noted a strong correlation between insulin resistance in the brain and early AD, suggesting that AD might be considered a neuroendocrine disorder of the brain or so-called “type 3 diabetes”[9, 10] .
• The association of AD with mitochondrial dysfunction [11].A genetic defect in mitochondrial Complex I genes is associated with a small minority (2%) of AD cases. It is noteworthy that mitochondrial defects, particularly in Complex I, have been found to be present in all major neurodegenerative diseases, not just AD, but also Parkinson's disease [12]and amyotrophic lateral sclerosis (ALS) [13,14].Mitochondrial dysfunction leads to two damaging conditions: insufficient ATP to fuel the cell's energy needs and oxidative damage due to excessive reactive oxygen species (ROS).
Senile or late onset Alzheimer’s disease
Neuronal apoptosis as more and more damage is incurred by the cell membranes, without sufficient replenishment of the supplies of fats and cholesterol to repair them, an increase in ion leakage across all membranes leads to further depletion of ATP and further exposure to pathogens and oxidative damage. Over time, the accumulated depletion of ATP leads to lysosomal dysfunction, likely because of an inability to maintain a sufficiently acidic pH for the digestive enzymes to work properly. In parallel, the further glycation and oxidation of Aβ convert it into an oligomeric complex with both decreased function and reduced susceptibility to degradation by the lysosomes. Once a sufficient percentage of the cell membrane has been compromised due to oxidative damage, it incurs rapid calcium influx and subsequent apoptosis. The protein plaques and tangles of AD are unrecyclable debris that remains in place after cell death.
Etiology of AD
1. Synthesis of acetylcholine is reduced in brain[15]
2. Amyloid beta (Aβ) deposited in brain
3. Access production of Apoliopoprotien E (Apo E) in brain by glial cells
4. TAU Protein
5. Inflammatory mediators
6. Degeneration of cholinergic neurotransmission
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Pathophysiologic Mechanisms of Neurological Pathways of AD
The pathophysiology of the AD involves the major alteration in brain. These alterations are of four types:
1. Cortical atrophy
2. Degeneration of cholinergic and other neurons
3. Presence of neuro-fibrillary tangles (NFTs)
4. Accumulation of neuritic plaques (NPs)
NFTs and neuritics plaques are considered the signature lesions of AD. In this two proteins are present i.e. beta amyloid protein and apolipoprotien E are contributed to the genesis of NFTs and Neuritics plaques.
1. Neuro fibrillary tangles (NFTs)
These are present in normal brain, their numbers increasing with age. In Alzheimer’s disease the quantity of these lesions drastically increased, particularly in the areas associated with memory and congnition, namely, the hippocampus, amygdale and cerebral cortex.
NFTs are located intracellularly, within the cytoplasm of neurons. NFTs are comprised of paired neurofilaments adopting a helical shape, unlike normal neurofilaments. The paired helical filaments is an abnormally phosphorylated from tau protein, a naturally occurring cell protein, commonly associated with microtubules. Microtubules are important in cellular transport. Abnormalities of tau protein and the presence of NFTs disrupt the cell structure, resulting in improper cell function and eventual cell death [16-18].
2. Neuritics plaques (Amyloid plaques/senile plaques)
The neuritic plaques differ from NFTs both in location and composition. Neuritic plaques are extracellular lesions found in the brain and cerebral vasculature. Plaques are comprised of a core of beta- amyloid protein, surrounded by a snarled mass of broken neuritis (axon and dendrite projections of neurons ). Many of these broken neuritis contain neuro-filaments made up of the abnormally phosphorylated tau protein found in NFTs. Also surrounded and entwined in the plaques are of two types of glial cells, astrocytes and microglia. Among other function, glial cells secrete inflammatory meditors and serve as scavengers’ cells, which may be important in considering inflammatory mechanism of Alzheimer’s disease. Neuritc plaques and NFTs significantally interfere with neuronal transmission pathway.
3. β –amyloid protein (β-AP)
β -AP are named for their beta pleated sheet structure, referring to the tertiary folding of the protein which is released from the Amyloid Precursor protein (APP) which is released from the secretases. Following release into the extrcellular space, β-AP protein fragments can adhere to one another, forming insoluble aggregates that are relatively resistant to destruction by prtoteases. As extracellular deposits of β –AP develop into mature neuritic plaques, they create physical barriers between nerve cells, which leads to alzheimers disease. Plaque formation seems to precede the accumulation of NFTs and neurofil threads, eventually coming nerve cell break down. Other evidence supports the theory that β –AP deposits initiate the wide spread structural damage associated with dementia [19-34] .
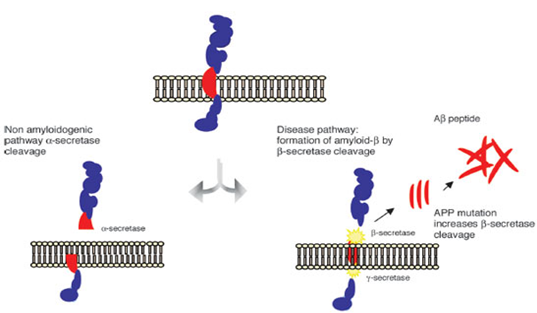
Figure1: The amyloid precursor protein (APP) is a transmembrane protein cleaved by secretase enzymes. In the non-amyloidogenic pathway, APP is cleaved preferentially by a-secretase. In the amyloidogenic pathway, neurotoxic Ab peptides are released after sequential cleavage of APP by b and g-secretases, and further accumulate into oligomeric aggregates. (Figure 1 reference: Vanessa de Jesus R. de Paula, Fabiana Meira Guimaraes, Breno Satler Diniz, Orestes Vicente Forlenza. Neurobiological pathways to Alzheimer's disease: amyloid-beta, Tau protein or both. Dementia Neuropsychologia 2009; vol. 3, 188-194.)
4. Apolipoprotein E (Apo E)
Apo E is well known for its role as a shuttle of cholesterol in blood. However, Apo E is also produced in the brain by glial cells, and its production is increased following injury of neuronal tissues. The genes responsible for the production of Apo E is located on chromosome-19 in a region previously noted to be associated with late onset Alzheimer’s disease. There are three major sub types or Iso forms of Apo E4. Apo E3 is the most common type (90% of individuals have at least one copy), with E2 and E4 occurring less frequently. Apo E2 protective in nature . Apo E3 is responsible for Alzheimer’s disease [35-47].
5. Tau Protein
The TAU protein is a microtubule-associated protein found in most tissues and which is highly expressed in the central and peripheral nervous system. It is an important component of the neuronal cytoskeleton, given its ability to interact with α- and β-globulin to stabilize the microtubules. In neurons, the microtubules are essential for the maintenance of neuronal structure, axonal transport, and synaptic plasticity.
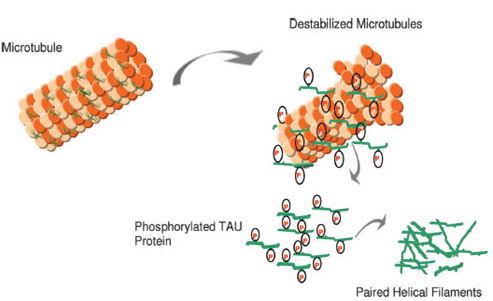
Figure2: In AD, there is a reduction in the ability to bind microtubules and promote microtubule assembly. Hyperphosphorylated Tau may contribute to a destabilized microtubule network, impaired axonal transport, and ultimately in neurofibrillary tangle (NFT) formation and neuronal death. (Figure 2 reference: Vanessa de Jesus R. de Paula, Fabiana Meira Guimaraes, Breno Satler Diniz, Orestes Vicente Forlenza. Neurobiological pathways to Alzheimer's disease: amyloid-beta, Tau protein or both. Dementia Neuropsychologia 2009; vol. 3, 188-194.)
Six TAU isoforms are defined in mammals. The main differences between these isoforms hinge on the existence of three or four binding domains to tubulin, in addition to some minor differences in the N-terminal end of the protein. The interaction between TAU and tubulin is a dynamic process in which TAU promotes its own polymerization and inhibits fast depolymerization of the tubulin. This process is regulated by the phosphorylation state of TAU protein, which comprises approximately 79 phosphorylation sites at serine and threonine residues. The balance between phosphorylation and dephosphorylation of these epitopes promotes conformational changes that influence how TAU protein interacts with α- and β-tubulin and that stabilize the microtubules in the neurons. Several protein kinases and phosphatases are involved in the regulation of TAU phosphorylation, the enzyme glycogen synthase kinase 3β (GSK3β) being the most important TAU-kinase in the neurons[48-57].
6. Inflammatory Mediators
Membrane attack complex(MAC) is found associated with broken neuritis and areas containing NFTs, implicating MAC as promoting the vast neuronal destruction characterising Alzheimer’s disease. Acute phase proteins α1-antichymotrypsin and α2- macro-globulins also act as a protease inhibitors, and could influence proteolytic break down of APP into β –AP [58, 59].
7. Degeneration of Cholinergic Neurotransmission
Cholinergic neurons are present in the hippocampus and amygdala. These areas are associated with higher learning and memory. In Alzheimer’s disease loss of cholinergic neurons occurs which leads to blockade of cholinergic neuro-transmission.
8. Others Neurotransmitter Abnormalities
Serotonergic neurons of the raphae nuclei and non-adrenergic cells of the locus cerulus are lost, while Mono Amine Oxidase type B (MAO-B) activity increased in Alzheimer’s disease. The presence of increased MAO-B concentration may seem counter intuitive, considering the vast neuronal loss in Alzheimer’s disease, unless one considers that MAO-B whose populations are increased. Other abnormalities also appears in the glutamate pathways, which is a major neurotransmitter in the cortex and hippocampus.
The Drug Treatment & Their Targets:
|
Drug Name |
Drug Type & Use |
Mechanism |
Recommended Dose |
|
Namenda (Memantine) |
N-methyl D-aspartate (NMDA) antagonist prescribed to treat symptoms of moderate to severe Alzheimer’s |
Blocks the toxic effects associated with excess glutamate and regulates glutamate activation |
Tablet and Oral Solution: Initial dose of 5 mg once a day May increase dose to 10 mg/day (5 mg twice a day), 15 mg/day (5 mg and 10 mg as separate doses), and 20 mg/day (10 mg twice a day) at minimum 1-week intervals if well tolerated. Extended Release Tablet: Initial dose of 7 mg once a day; may increase dose to 14 mg/day, 21 mg/day, and 28 mg/day at minimum 1-week intervals if well tolerated. |
|
Razadyne (Galantamine) |
Cholinesterase inhibitor prescribed to treat symptoms of mild to moderate Alzheimer’s |
Prevents the breakdown of acetylcholine and stimulates nicotinic receptors to release more acetylcholine in the brain |
Tablet & Oral solution: Initial dose of 8 mg/day (4 mg twice a day) May increase dose to 16 mg/day (8 mg twice a day) and 24 mg/day (12 mg twice a day) at minimum 4-week intervals if well tolerated Extended Release capsule: The dosage is same as tablet and oral solution but they are taken once a day |
|
Exelon (Rivastigmine) |
Cholinesterase inhibitor prescribed to treat symptoms of mild to moderate Alzheimer’s |
Prevents the breakdown of acetylcholine and butyrylcholine (a brain chemical similar to acetylcholine) in the brain |
Capsule & Oral solution: Initial dose of 3 mg/day (1.5 mg twice a day) May increase dose to 6 mg/day (3 mg twice a day), 9 mg (4.5 mg twice a day), and 12 mg/day (6 mg twice a day) at minimum 2-week intervals if well tolerated/. Patch: Initial dose of 4.6 mg once a day; may increase to 9.5 mg once a day after minimum of 4 weeks if well tolerated. |
|
Aricept (Donepezil) |
Cholinesterase inhibitor prescribed to treat symptoms of mild to moderate, and moderate to severe Alzheimer’s |
Prevents the breakdown of acetylcholine in the brain |
Tablet: Initial dose of 5 mg once a day May increase dose to 10 mg/day after 4-6 weeks if well tolerated, then to 23 mg/day after at least 3 months Orally Disintegrating Tablet: The dose of the orally disintegrating tablet is same as the doses of the tablet. 23-mg dose available as brand-name tablet only |
Table ref.:
Alzheimer's Disease Education and Referral (ADEAR) Center, A Service of the National Institute on Aging National Institutes of Health, U.S. Department Of Health And Human Services , November 2008 Updated July 2010 NIH Publication No. 08-3431.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Conclusion
Alzheimer disease (AD) is the most common causes of neurodegenerative disorder in the elderly individuals. Clinically, patients initially present with short-term memory loss, subsequently followed by executive dysfunction, confusion, agitation, and behavioural disturbances. The present review study of neurological pathways shows that these all pathways will leads to the AD. In this review article, we also provide an overview of drug treatment of AD according to their target site.
References
1. Herrera E Jr, Caramelli P, Silveira AS, Nitrini R. Epidemiologic survey of dementia in a community-dwelling Brazilian population. Alzheimer Dis Assoc Disord 2002; 16:103-108.
2. Nitrini R, Caramelli P, Herrera E Jr, et al. Incidence of dementia in a community-dwelling Brazilian population. Alzheimer Dis Assoc Disord 2004; 18: 241-246.
3. Hestad K, Kveberg B, Engedal K. Low blood pressure is a better predictor of cognitive deficits than the apolipoprotein e4 allele in the oldest old. Acta Neurol Scand 2005; 111:323-328.
4. Ravona SR, Davidson M, Noy S. The role of cardiovascular risk factors in Alzheimer's disease. CNS Spectr 2003; 8: 824-833.
5. Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H. Alzheimer's presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging 2008; 29:319-328.
6. Castellani RJ, Lee HG, Zhu XW, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic, Acta Neuropathol 2006; 111:503-509.
7. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239-259.
8. Berchtold NC, Cotman CW. "Evolution in the conceptualization of dementia and Alzheimer's disease: Greco-Roman period to the 1960s". Neurobiol. Aging 1998; 19 (3): 173–89.
9. Schubert D. Glucose metabolism and Alzheimer's disease. Ageing Res Rev 2005; 4: 240-57.
10. Steen E, Terry BM, Rivera EJ, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease—is this type 3 diabetes? J Alzheimers Dis 2005; 7:63–80.
11. Alikhani N, Ankarcrona M, Glaser E. Mitochondria and Alzheimer's disease: amyloid-β peptide uptake and degradation by the presequence protease, hPreP. J Bioenerg Biomembr 2009; 41:447–51.
12. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 1990; 54:823–7.
13. Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 2005;5:77–87.
14. Dawson VL. Maiming mitochondria in familial ALS. Nat Med 2004; 10:905–6.
15. Francis PT, Palmer AM, Snape M, Wilcock GK (February 1999). “The cholinergic hypothesis of alzheimer’s disease: a review of progress”. J. Neurol. Neurosurg. Psychiatr. 1999; 66 (2): 137–47.
16. Selkoe, D. J., Ihara, Y. & Salazar, F. J. Science.1982; 215, 1243-1245.
17. Ignatius, M. J., Gebicke-Harter, P. J., Skene, J. H. P., Schilling,J. W., Weisgraber, K. H., Mahley, R. W. & Shooter,E. M. Proc. Natl. Acad. Sci. USA. 1986; 83, 1125-1129.
18. Pitas, R. E., Boyles, J. K., Lee, S. H., Foss, D. & Mahley,R. W. (1987) Biochim. Biophys. Acta. 1987; 917, 148-161.
19. Merz, P. A., Somerville, R. A., Wisniewski, H. M. & Iqbal, K. Acta Neuropathol. 1981; 54, 63-74.
20. Ravona SR, Davidson M, Noy S. “The role of cardiovascular risk factors in Alzheimer's disease”. CNS Spectr 2003; 8:824-833.
21. Mahley, R. W. Science.1988; 240, 622-628.
22. Boyles, J., Pitas, R. E., Wilson, E., Mahley, R. W. & Taylor,J. M. J. Clin. Invest. 1985; 76, 1501-1513.
23. Elshourbagy, N. A., Liao, W. S., Mahley, R. W. & Taylor,J. M. Proc. Natl. Acad. Sci. USA. 1985; 82, 203-207.
24. Boyles, J. K., Zoellner, C. D., Anderson, L. J., Kosik, L. M.,Pitas, R. E., Weisgraber, K. H., Hui, D. Y., Mahley, R. W., Gebicke-Haerter, P. J., Ignatius, M. J. & Shooter, E. M. J. Clin. Invest.1989; 83, 1015-1031.
25. Snipes, G. J., McGuire, C. B., Norden, J. J. & Freeman, J. A. Proc. Nat!. Acad. Sci. USA. 1986; 83, 1130-1134.
26. Namba, Y., Tomonaga, M., Kawasaki, H., Otomo, E. & Ikeda,K. Brain Res. 1991; 541, 163-166.
27. Jiang Q, Lee CYD, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Aβ. Neuron 2008; 58:681–93.
28. Koistinaho M, Lin S,Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med 2004;10: 719–26.
29. Bishop T, St-Pierre J, Brand MD. Primary causes of decreased mitochondrial oxygen consumption during metabolic depression in snail cells. Am J Physiol Regul Integr Comp Physiol 2002; 282:R372–82.
30. Zhao W, De Felice FG, Fernandez S, et al. Amyloid-β oligomers induce impairment of neuronal insulin receptors. FASEB J 2008;22: 246–60.
31. Miller BC, Eckman EA, Sambamurti K, et al. Amyloid-β peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA 2003; 100:6221–6.
32. Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A β-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol 2001;281: H2417–24.
33. Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid β-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci 1997; 17: 1046–54.
34. Yan SD, Roher A, Chaney M, Zlokovic B, Schmidt AM, Stern D. Cellular cofactors potentiating induction of stress and cytotoxicity by amyloid β-peptide. Biochim Biophys Acta 2000; 1502:145–57.
35. Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol 2001; 12:105–12.
36. Pfrieger FW. Role of cholesterol in synapse formation and function. Biochim Biophys Acta 2003; 1610:271–80.
37. Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med 1996; 47:387–400.
38. Pitas RE, Boyles JK, Lee SH, Foss D, Maley RW. Astrocytes synthesize apolipoprotein E and metabolize apoliporotein E-containing lipoproteins. Biochem Biophys Acta1987; 917:148–61.
39. DeMattos RB, Brendza RP, Heuser JE, et al. Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice. Neurochem Int 2001; 39:415–25.
40. Rapp A, Gmeiner B, Huttinger M. Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 2006; 88:473–83.
41. Notkola IL, Sulkava R, Pekkanen J, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology 1998; 17:14–20.
42. West R, Beeri MS, Schmeidler J, et al. Better memory functioning associated with higher total and low-density lipoprotein cholesterol levels in very elderly subjects without the apolipoprotein e4 allele. Am J Geriatr Psychiatry 2008; 16:781–5.
43. Mielke MM, Zandi PP, Sjgren M, et al. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology 2005; 64:1689–95.
44. Mulder M, Ravid R, Swaab DF, et al. Reduced levels of cholesterol, phospholipids, and fatty acids in cerebrospinal fluid of Alzheimer disease patients are not related to apolipoprotein E4. Alzheimer Dis Assoc Disord 1998; 12:198–203.
45. Honjo K, van Reekum R, Verhoeff NPLG. Alzheimer's disease and infection: do infectious agents contribute to progression of Alzheimer's disease? Alzheimers Dement 2009; 5:348–60.
46. Dimopoulos N, Piperi C, Salonicioti A, et al. Characterization of the lipid profile in dementia and depression in the elderly. J Geriatr Psychiatry Neurol 2007; 20:138–44.
47. Michikawa M, Yanagisawa K. Apolipoprotein E4 induces neuronal cell death under conditions of suppressed de novo cholesterol synthesis. J Neurosci Res 1998; 54:58–67.
48. Kosik KS. The molecular and cellular biology for tau. Brain Path 1993; 3:39-43.
49. Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 1984; 259:5301-5305.
50. Cleveland DW, Hoffman PN. Neuronal and glial cytoskeletons. Curr Opin Neurobiol 1991; 1:346-353.
51. Lovestone S, Anderton B. Cytoskeletal abnormalities in Alzheimer's disease. Curr Opin Neurol Neurosurg 1992; 5:883-888.
52. Trojanowski JQ, Lee VM. Paired helical filament tau in Alzheimer's disease. The kinase connection: neurobiology of Alzheimer's disease. Am J Pathol 1994; 144:449-453.
53. Lovestone S, Reynolds CH, Latimer D, et al. Alzheimer's disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol 1994; 4:1077-1086.
54. Shahani N, Brandt R. Functions and malfunctions of the tau proteins. Cell Mol Life Sci.2002; 39:1668-1680.
55. Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 1992; 3:1141-1154.
56. Hernández F, Avila J. Tauopathies. Cell Mol Life Sci 2007; 64:2219-2233.
57. Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Rev Brain Res 2000; 33:95-130.
58. Wenk GL (2003). "Neuropathologic changes in Alzheimer's disease". J Clin Psychiatry, 2003; 64 Suppl 9: 7–10.
59. Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP. (2010). Locus ceruleus controls Alzheimer’s Disease pathology by modulationg microglial funcation through norepinephrine. Proc Natl Acad Sci U S A. 2010; 107:6058–6063.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






